EPIDEMIOLOGY AND ETIOLOGY
Acute lymphoblastic leukemia (ALL) is characterized by the proliferation and accumulation of lymphoid progenitor cells in the blood, bone marrow, and other tissues. It has a bimodal distribution. The overall age-adjusted incidence is 1.7 per 100,000 persons, but ALL affects 4 to 5 per 100,000 persons during age 4 to 5 years and half that number around the fifth decade of life. Approximately 60% of cases are diagnosed in patients ≤20 years old, with a median age at diagnosis of 14 years. In 2014, the American Cancer Society estimated that approximately 6,000 individuals would be diagnosed with ALL that year (1,2). Acute lymphoblastic leukemia represents 20% of adult leukemias but is the most common childhood acute leukemia, representing approximately 80% of cases (1,2).
The etiology of ALL is unknown in most cases (3,4,5,6,7). Chromosomal translocations occurring in utero during fetal hematopoiesis have suggested genetic factors as the primary cause for pediatric ALL and postnatal genetic events as secondary contributors. Monozygotic and dizygotic twins of patients with ALL and individuals with genetic disorders, such as Klinefelter (XXY and variants) and Down (trisomy 21) syndromes, or inherited diseases with excessive chromosomal fragility, such as Bloom syndrome, Fanconi anemia, and ataxia telangiectasia, have all been found to have higher incidence of ALL, implicating a possible genetic predisposition. Additional studies have postulated infectious etiologies (4). Human T-cell lymphotropic virus type-1 is known to cause adult T-cell leukemia/lymphoma (5); Epstein-Barr virus has been associated with lymphoproliferative disorders, including Burkitt lymphoma and mature B-cell ALL (6); and varicella has been linked to childhood ALL (7).
CLINICAL PRESENTATION AND LABORATORY ABNORMALITIES
Presenting symptoms can be nonspecific, particularly in children. They largely reflect bone marrow failure and include malaise, fatigue, bleeding or bruising, and secondary infections. The B symptoms, such as fever, night sweats, and weight loss, are frequent. White blood cell (WBC) count at presentation varies widely, and circulating blasts are generally noted. Symptoms related to hyperleukocytosis are rare in ALL, given the lymphoblast morphology, even when WBC counts are high.
Leukemic involvement of the central nervous system (CNS) ranging from cranial neuropathies to meningeal infiltration occurs in <10% of patients at presentation. It is more common in mature B-cell ALL (Burkitt leukemia) (8). A history or findings of abdominal masses, significant spontaneous tumor lysis syndrome, and chin numbness (mental nerve) indicating cranial nerve involvement are also more common in this subtype of ALL (9). Lymphadenopathy and hepatosplenomegaly, although rarely symptomatic, are noted in approximately 20% of patients (9).
DIAGNOSIS
The diagnosis of ALL is largely based on flow cytometric immunophenotyping, although identification of cytogenetic-molecular abnormalities plays a significant role (Fig. 1-1). The World Health Organization (WHO) proposed new guidelines for the diagnosis of neoplastic diseases of hematopoietic and lymphoid tissues (10). The French-American-British (FAB) Cooperative Group diagnostic approach, which recognizes L1 to L3 morphologic subtypes, has been essentially abandoned. A blast count of ≥20% was established as sufficient for diagnosis.
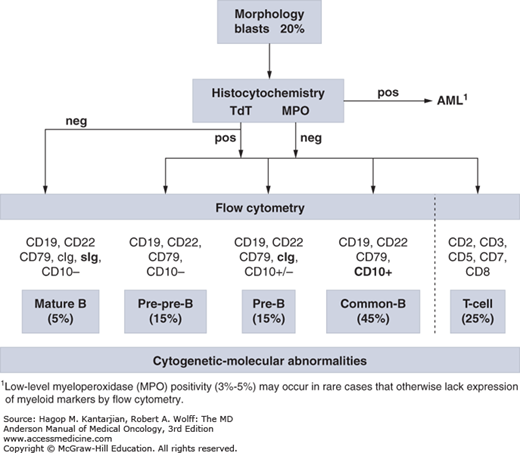
Flow cytometric analysis successfully assigns lineage in more than 95% of cases. True mixed phenotype acute leukemia is rare (11). Concomitant expression of markers from more than one lineage is seen in 15% to 50% of adult and 5% to 35% of pediatric ALL (12,13,14), but this is not prognostically relevant. Targeted genomic profiling may further define ALL subtypes with different response profiles to therapy and prognoses, which are only partially discriminated by current diagnostic tools.
Immunophenotypically, ALL blasts are negative for myeloperoxidase (MPO), although low-level MPO positivity (3%-5%) may occur in rare cases that otherwise lack expression of myeloid markers by flow cytometry (15). Terminal deoxynucleotidyl transferase (TdT), although not a specific marker of ALL, helps separate malignant lymphocytosis from reactive processes and distinguish L3 ALL (TdT negative) from other ALL subtypes (16).
Both the prior FAB and current WHO classification systems rely heavily on morphologic assessment (17), which accounts for cell size, cytoplasm, nucleoli, basophilia, and vacuolation. The former FAB L3 morphology, characterized by a high rate of cell turnover, is associated with mature B-cell ALL (Burkitt leukemia) and gives rise to the “starry sky” pattern on marrow biopsies.
Three broad immunophenotypic ALL groups can be distinguished: precursor B-cell, mature B-cell, and T-cell ALL (Table 1-1). Precursor B-cell ALL (B-ALL) stains positive for TdT, HLA-DR, CD19, and CD79a. According to the stages of maturation, further B-cell subgroups have been defined as pre-pre-B-ALL (pro–B-ALL), common ALL, and pre–B-ALL. Although they all stain positive for CD19, CD79a, or CD22, expression of CD10 (common ALL antigen [CALLA]) distinguishes common ALL (early pre–B-ALL), and cytoplasmic immunoglobulins with or without CD10 identify pre–B-ALL. Mature B-ALL (Burkitt leukemia) is TdT negative but expresses surface immunoglobulins (usually immunoglobulin M), as well as κ or λ light chains in a clonal fashion. It has almost ubiquitous expression of CD20, which has therapeutic implications (18).
| B Lineage | T Lineage | ||
|---|---|---|---|
| CD19/CD79a/CD22 | CD3 (Surface/Cytoplasmic) | ||
| Pre-pre-B-ALL (pro–B-ALL) | — | Precursor T-ALL | CD1a, CD2, CD5, CD7, CD8, cCD3 |
| Common ALL | CD10 (CALLA) | Mature T-ALL | Surface CD3 (plus any other T-cell markers) |
| Pre–B-ALL | Cytoplasmic IgM | ||
| Mature B-ALL | Cytoplasmic or surface Ig κ or λ | ||
T-cell ALL (T-ALL) further stratifies into subtypes based on different stages of thymic differentiation (19). As the most lineage-specific marker for T-cell differentiation, surface CD3 (sCD3) is typically positive in mature T-ALL, which is also positive for either CD4 or CD8, but not both. However, pre–T-ALL is negative for CD4, CD8, and sCD3 but may still express cytoplasmic CD3. A more simplified classification divides T-ALL into early T-ALL (sCD3–, CD1a–), thymic T-ALL (sCD3+/–, CD1a+), and mature T-ALL (sCD3+, CD1a+). Only thymic T-ALL has excellent outcome with chemotherapy alone.
Frequent cytogenetic and molecular abnormalities associated with adult ALL offer insight into the events leading to leukemic progression (Table 1-2) (20). They are of both prognostic and predictive significance and have varying frequencies in children and adults, which explains some of the differences in outcomes in these two groups. This is particularly true in the case of ALL harboring Philadelphia chromosome [t(9;22)] (Ph) or other chromosomal changes with prognostic relevance such as Burkitt karyotypes [t(8;14), t(2;8), t(8;22)] or t(4;11). Next-generation sequencing, expression proteomics, and oligonucleotide microarrays have transformed our understanding of the genomic landscape of ALL and are yielding new molecular subgroups with actionable defects (21,22,23).
| Category | Cytogenetics | Involved Genes | Adults Frequency (%) | Children Frequency (%) |
|---|---|---|---|---|
| Hyperdiploid | 2-15 | 10-26 | ||
| Hypodiploid | 5-10 | 5-10 | ||
| Pseudodiploid | t(9;22)(q34;q11) | BCR-ABL1 | 15-25 | 2-6 |
| del(9)(q21-22) | p15, p16 | 6-30 | 20 | |
| t(4;11);t(9;11); | MLL | 5-10 | <5 | |
| t(11;19); t(3;11) | ||||
| del(11)(q22-23) | ATM | 25-30a | 15a | |
| t(12;21)(p12;q22) | TEL-AML1 | <1b | 20-25b | |
| t(1;19) | E2A-PBX1 | <5 | <5 | |
| t(17;19) | E2A-HLF | <5 | <5 | |
| t(1;14)(p32;q11) | TAL1 | 10-15 | 5-10 | |
| t(7;9)(q34;q32) | TAL2 | <1 | <1 | |
| t(10;14)(q24;q11) | HOX11 | 5-10 | <5 | |
| t(5;14)(q35;q32) | HOX11L2 | 1 | 2-3 | |
| t(1;14)(p32;q11) | TCR | 20-25c | 20-25c | |
| del(13)(q14) | miR15/miR16 | <5 | <5 | |
| t(8;14); t(8;22); t(2;8) | C-MYC | 5 | 2-5 | |
| +8 | ? | 10-12 | 2 | |
| del(7p) | ? | 5-10 | <5 | |
| del(5q) | ? | <2 | <2 | |
| del(6q); t(6;12) | ? | 5 | <5 |
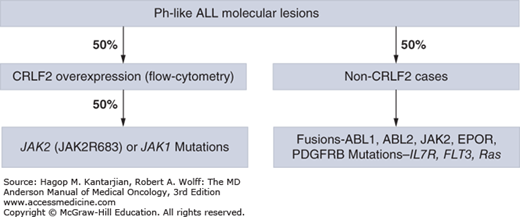
Recently, a Ph-like signature in 10% of children with standard-risk ALL and as many as 25% to 30% of young adults with ALL has been defined using genome-wide gene expression arrays. This subgroup lacks the expression of BCR-ABL1 fusion protein but does have a gene expression profile similar to BCR-ABL1 ALL (24,25,26). The vast majority of these patients have deletions in key transcription factors involved in B-cell signaling, such as IKZF1, TCF3, EBF1, PAX5, and VPREB1, as well as kinase-activating alterations involving ABL1, ABL2, CRLF2, CSF1R, EPOR, JAK2, NTRK3, PDGFRB, PTK2B, TSLP, or TYK2 and sequence mutations involving FLT3, IL7R, or SH2B3. The most common alterations (~50%) are rearrangements of CRLF2, which activate downstream signaling through JAK kinases, and approximately half of these cases have activating mutations in JAK1 or JAK2 (Fig. 1-2). Importantly, patients with ABL1, ABL2, CSF1R, and PDGFRB expression fusions were sensitive in in vitro and in vivo human xenograft models to ABL class tyrosine kinase inhibitors (TKIs; eg, dasatinib); rearrangements in EPOR, IL-7R, and JAK2 mutations and fusions were sensitive to JAK kinase inhibitors (eg, ruxolitinib); and patients with ETV6-NTRK3 fusion were sensitive to ALK kinase inhibitors (eg, crizotinib) (25), further expanding therapeutic options in this subgroup with poor outcome.
Observations of epigenetic alterations regulating distinct molecular pathways that occur frequently at presentation and relapse have identified a “hypermethylator” phenotype of ALL (27). These patients may respond favorably to treatment with hypomethylating agents (azacitidine or decitabine). Identification of these and other molecular and cytogenetic changes in adult ALL drives the development of risk-adapted and targeted therapies, particularly in high-risk groups (Table 1-3) (28).
| ALL Lineage | Cytogenetic Aberration | Involved Genes | Protein | Comments |
|---|---|---|---|---|
| B cell | BCR/ABL+ (Ph+) | IKZF1 | Ikaros | Poor outcome. 80% of Ph+ cases. |
| CRLF2 + the Ig heavy chain locus; or an interstitial PAR1 deletion | CRLF2 | 5%-10% of cases with no molecular rearrangement. Poor outcome. 50% of children with Down syndrome. | ||
| BCR/ABL-like | IKZF1 deletions; rearrangements/mutations in CRLF2, IGH–CRLF2, and NUP214–ABL1; in-frame fusions of EBF1-PDGFRB, BCR-JAK2, or STRN3-JAK2; cryptic IGH-EPOR rearrangements | 15% of cases. Potential use of TKIs and/or mTOR and JAK2 inhibitors. | ||
| Near hypodiploid | NRAS, KRAS, FLT3, and NF1 | 70% of cases. | ||
| Low hypodiploid | IKZF2, and by TP53 disruptions, CDKN2A/B locus deletion | 91% of cases. | ||
| Hyperdiploid | CREBBP | |||
| NT5C2 mutations | NT5C2 | |||
| TP53 mutations | 6% of cases. | |||
| T cell | PICALM-MLLT10, NUP214-ABL1 fusion, EML-ABL1, SET-NUP214 fusion, MLL, NOTCH1, FBW7, BCL11B, JAK1, PTPN2, IL7R, PHF6, RAS/PTEN | NOTCH1 (>60%) and/or FBW7 (~20%) mutations associated with a favorable outcome. RAS/PTEN and JAK1 usually poor outcome. |
FRONTLINE THERAPY
Therapy for ALL consists of complex and comprehensive regimens consisting of several phases: induction, intensified consolidation, maintenance, and CNS prophylaxis (9,29). Each involves the use of a core group of agents considered the backbone of therapy in a time- and dose-dependent manner, with a goal of restoring normal hematopoiesis, eradicating resistant subclones, providing adequate prophylaxis of sanctuary sites (eg, CNS, testicles), and eliminating minimal residual disease (MRD) during the consolidation and maintenance phases (9,30). Combining anthracyclines (eg, daunorubicin or doxorubicin), vincristine, and dexamethasone (for better CNS penetration), often coupled with cyclophosphamide or asparaginase with growth factor support, represents the cornerstone of ALL induction regimens. This results in complete remission (CR) rates of 70% to 90% and median remission durations of 18 months (30,31). Patients who achieve CR subsequently transition to the consolidation phase, which, depending on the risk-oriented subtype, may consist of consolidation chemotherapy (cytarabine, methotrexate, cyclophosphamide, and 6-mercaptopurine) or allogeneic hematopoietic stem-cell transplantation (AHSCT). Consolidation is followed by prolonged maintenance therapy with daily 6-mercaptopurine, weekly methotrexate, and monthly pulses of vincristine and prednisone or dexamethasone, given over 2 to 3 years (POMP or DOMP, depending on corticosteroid used) (30,31,32). Maintenance, which is omitted in mature B-ALL due to high cure rates, may also involve the use of TKIs for patients with Ph-positive ALL. Primary CNS involvement at diagnosis is rare (<10%) but is as high as 50% to 75% at 1 year without prophylactic administration of intrathecal chemotherapy (IT) (31). Although high-dose cytarabine (1-7.5 mg/m2) and methotrexate (5-8 g/m2) successfully penetrate the blood-brain barrier, they are too toxic to serve as the sole CNS prophylaxis. The inclusion of IT prophylaxis (methotrexate, cytarabine, liposomal cytarabine, hydrocortisone, or thiotepa) reduces the incidence of CNS relapse to 4% by allowing sustained therapeutic concentration of the agents in the cerebrospinal fluid. The number of ITs varies according to protocol (usually 8 for standard risk, 12 for Ph positive, and 16 for Burkitt), and in rare cases of extramedullary disease spread (eg, masses or chloromas), IT may even be supplemented by radiation therapy.
One extensively studied regimen used in treatment of adult ALL is the hyper-CVAD (HCVAD) regimen, where patients receive hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with high-dose methotrexate and cytarabine for a total of eight alternating cycles approximately every 3 to 4 weeks (Table 1-4) (30,31). This is followed by 2 years of POMP maintenance therapy, interspersed with intensification courses during months 6, 7, 18, and 19. The number of IT injections (two per course) depends on the risk of CNS relapse, which has been identified as high for patients with mature B-ALL. Our current approach is giving 8 ITs for nonmature B-ALL and 16 ITs for mature B-ALL, resulting in a 5-year overall survival (OS) between 38% and 50% (30). Due the improved cure rates of Ph-positive ALL patients, an increase in the CNS relapse rate was observed, which is the reason the protocol was modified to include 12 ITs for Ph-positive ALL.
| Therapy Segment | Dose and Schedule |
|---|---|
| Induction and intensified consolidation | Hyper-CVAD (courses 1, 3, 5, and 7) |
| • Cyclophosphamide 300 mg/m2 IV over 3 h every 12 h for 6 doses on days 1-3 | |
| • Mesna 600 mg/m2 as an IV continuous infusion over 24 h daily on days 1-3 (starting approximately 1 h prior to cyclophosphamide and finishing 12 h after the last dose) | |
| • Doxorubicin 50 mg/m2 IV continuous infusion over 24 h on day 4 | |
| • Vincristine 2 mg IV on days 4 and 11 | |
| • Dexamethasone 40 mg daily on days 1-4 and 4-11 | |
| Methotrexate (MTX) and high-dose cytarabine (courses 2, 4, 6, and 8) | |
| • MTX 200 mg/m2 IV over 2 h followed by 800 mg/m2 IV over 22 h on day 1 | |
| • Leucovorin rescue 15 mg every 6 h for eight doses (starting 12 h after completion of MTX) | |
| • Cytarabine 3 g/m2 IV over 2 h every 12 h for 4 doses on days 2 and 3 | |
| • Methylprednisolone 50 mg IV twice daily on days 1-3 | |
| CNS prophylaxis | IT MTX 12 mg (6 mg if via Omaya reservoir) on day 2 and cytarabine 100 mg on day 7 of each course |
| Low risk: 6 IT | |
| High risk: 8 IT | |
| Mature B cell: 16 IT | |
| Maintenance therapy | POMP |
| • 6-Mercaptopurine 50 mg orally three times per day | |
| • MTX 20 mg/m2 orally weekly | |
| • Prednisone 200 mg orally days 1-5 every month | |
| • Vincristine 2 mg IV every month | |
| • Intensification with four additional courses of hyper-CVAD plus MTX/cytarabine | |
| Supportive care | • Antibiotic prophylaxis (levofloxacin, fluconazole, valacyclovir) |
| • Hematopoietic growth factor support during induction and consolidation | |
| • Laminar air flow rooms (for patients ≥60 years old) |
The addition of rituximab to short intensive chemotherapy has also improved outcome in adult Burkitt and Burkitt-type lymphoma or ALL (29,33,34). Hoelzer and colleagues have recently reported the benefit of adding rituximab to short intensive chemotherapy in 363 patients with Burkitt lymphoma/leukemia; the addition of rituximab resulted in CR and 5-year survival rates of 88% and 80%, respectively (33). Higher rates of survival were reported in adolescents compared to adults and elderly patients (90% vs 84% vs 62%, respectively) (33). Low-intensity chemotherapy with infused etoposide, doxorubicin, and cyclophosphamide with vincristine, prednisone, and rituximab (EPOCH-R) was recently tested in 30 adult patients with Burkitt lymphoma (35). The progression-free survival (PFS) and OS rates were 90% and 100%, respectively. Of note, marrow involvement was present in only 13% of patients, and CNS involvement was present in only 3% of patients (35).
There have been several alterations to traditional protocols with further refining of the disease. Expression of cell surface marker CD20 in adult ALL ranges from 35% to ubiquitous depending on the subtype and has been associated with an inferior prognosis (18). The addition of two doses of monoclonal CD20 antibody (rituximab) administered with the first four cycles of chemotherapy and during maintenance intensification at months 6 and 18 resulted in improved OS in younger patients compared with similar chemotherapy historical controls (75% vs 47% at 3 years; P = .003) (36). Improvement in the 5-year remission duration and survival rates was also reported in patients <55 years old by the German Multicenter Study Group for ALL (GMALL) when rituximab was added to standard induction and consolidation therapy (37).
Ofatumumab is a more potent second-generation anti-CD20 monoclonal antibody that binds to a membrane proximal small-loop epitope on the CD20 protein. A phase II study in CD20-positive pre–B-ALL combined ofatumumab with HCVAD during induction, resulting in a 96% rate of both CR and MRD negativity. At a median follow-up of 14 months, the 1-year PFS and OS rates were 94% and 92%, respectively (38).