Children with Down syndrome have an increased risk for developing both acute myeloid as well as lymphoblastic leukemia. These leukemias differ in presenting characteristics and underlying biology when compared with leukemias occurring in non-Down syndrome children. Myeloid leukemia in children with Down syndrome is preceded by a preleukemic clone (transient leukemia or transient myeloproliferative disorder), which may disappear spontaneously, but may also need treatment in case of severe symptoms. Twenty percent of children with transient leukemia subsequently develop myeloid leukemia. This transition offers a unique model to study the stepwise development of leukemia and of gene dosage effects mediated by aneuploidy.
Childhood leukemias often originate from a premalignant clone, a generally asymptomatic condition that can only be detected by demonstrating leukemia-specific genetic changes in peripheral blood, including that obtained from Guthrie cards or in cord-blood. Subsequently, secondary genetic events cause outgrowth of this dormant clone and result in frank leukemia. However, the likelihood of developing leukemia in children who carry premalignant clones, harboring genetic aberrations such as TEL-AML or AML1-ETO is low. Because of the very low transformation into frank leukemia, prevention strategies are generally considered not feasible.
Children with Down syndrome (DS) (for a review on DS, see Roizen and Patterson in ) have an increased risk of developing leukemias, which was already recognized in the 1950s, although their risk of cancer in general is not increased because of a reduced propensity for solid tumors. This increased risk of leukemia includes both acute lymphoblastic leukemia as well as myeloid leukemia. Approximately 10% of newborns with DS develop a preleukemic clone, originating from myeloid progenitors in the fetal liver that are characterized by a somatic mutation in the gene encoding for the hematopoietic transcription factor GATA1, which is localized on the X-chromosome. Mutations in this transcription factor lead to a truncated mutant protein GATA1short or GATA1s. This preleukemia is referred to as transient leukemia (TL), transient myeloproliferative disease (TMD), or transient abnormal myelopoiesis (TAM).
Transient leukemia is associated with a variable clinical presentation, ranging from a symptomatic to severe complications, which may even be fatal. As TL originates in the liver, peripheral blood blast counts are usually higher than the bone marrow blast count. Subsequently, approximately 20% of children who have been diagnosed with TL develop myeloid leukemia of Down syndrome (ML DS; this term has been introduced to refer to the unique subtype of AML that develops in children with DS, as explained later in this article), usually with an onset before the age of 5 years. Studying TL and ML DS may contribute significantly to our understanding of the stepwise process of leukemogeneis. However, so far the genetic factors that drive this progression are unknown. Mutations in the GATA1 gene are present both in both TL and ML DS, and are therefore not sufficient to explain this progression.
Of interest, children with DS are also predisposed to develop acute lymphoblastic leukemia (ALL), which is not associated with an overt preleukemic phase. Both ML and ALL in patients with DS are biologically distinct when compared with their non-DS counterparts, and are characterized by a different clinical behavior. For children with DS who develop ALL, this does not lead to improved outcome, as is observed for children with DS who develop ML. This article reviews the current knowledge and research questions regarding TL and the DS-associated acute leukemias.
Hematologic abnormalities in children with Down syndrome
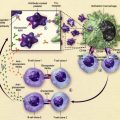
Tunstall-Pedoe and colleagues studied hematopoiesis in fetal bood, bone marrow, and liver cells from 16 fetuses with DS with a gestational age of 15 to 37 weeks. GATA1 mutations were not detected in the hematopoietic cells of these fetuses, although minor clones may have been missed, as explained later in this article. A marked increase in the megakaryocyte-erythrocyte progenitors in the fetal liver was found, as well as dysmegakaryopoiesis and dyserythropoiesis in the peripheral blood, but not in the bone marrow, when compared with non-DS fetuses. The investigators conclude that trisomy 21 by itself affects fetal hematopoiesis, and that the expansion of fetal liver progenitors creates a cellular substrate for GATA1 mutations to occur, which subsequently gives rise to TL (see later in this article and Fig. 1 ).
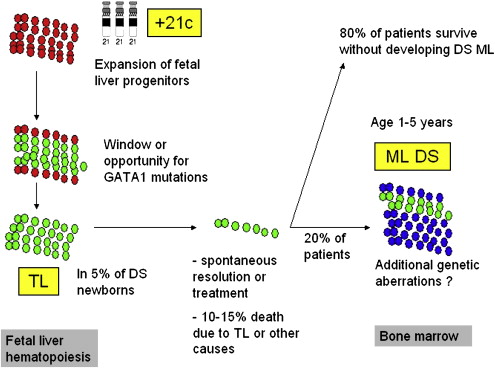
In DS newborns (n = 158), peripheral blood abnormalities in the first week of life included neutrophilia in 80% of children, thrombocytopenia in 66% (6% had platelets less than 50 × 10 9 /l) and polycythemia in 33%. Anemia, neutropenia, and thrombocytosis were present in less than 1% of DS newborns. In 6% of children, TL was diagnosed. Widness and colleagues suggested that the observed polycythemia may be caused by increased erythopoietin levels when compared with non-DS newborns.
After the first week of life, the main peripheral blood abnormalities consist of red blood cell macrocytosis and thrombocytosis, although this study was limited to DS children who were younger than 1 year of age.
De Hingh and colleagues have studied T- and B-lymphocyte counts in children with DS and compared these to children without trisomy 21. Over time, the T-lymphocyte subpopulation gradually reached normal levels, but the B-lymphocyte population appeared severely decreased (88% of values were below the tenth percentile, and 61% below the fifth percentile of normal). These abnormalities may at least in part explain the increased susceptibility to infections, which children with DS may experience.
How frequent is transient leukemia in Down syndrome children?
The frequency of TL is estimated to be around 10% in DS children, although this was studied in a selected (hospitalized) population. Most investigators assume that the true frequency is higher, but recent data suggest that his may not be the case. In The Netherlands, children with DS are registered in different nation-wide registries (the Dutch Pediatric Surveillance Unit and the National Dutch Neonatal and Obstetrical Registry), which provide population-based data. After matching these databases, it was assessed that 322 children (95% confidence interval 303–341) with DS were born in 2003, resulting in a prevalence of 16 out of 10,000 live-born babies. To date, five children with DS who were born in 2002 and 2003 have been diagnosed with myeloid leukemia (by the Dutch Childhood Oncology Group, E.R. van Wering, personal communication, 2007). As ML in patients with DS typically occurs before the age of 5 years, with a median age of approximately 2 years, and the frequency for progression from TL to ML DS is well established at 20%, these data suggest that the true frequency of TL is in the 3% to 5% range.
This is further supported by a recent study from Pine and colleagues, who screened Guthrie cards from 590 infants with DS for mutations in the GATA1 gene and found a mutation in 3.8% of children. However, GATA1 mutations may have been missed in those patients with minor preleukemic clones, subclonal mutations, low numbers of cells on Guthrie cards, or extramedullary TL without circulating blasts. In addition, one has to consider that some fetuses with DS may die in utero from causes that may include TL. Prospective population-based studies regarding the incidence of TL are currently underway in The Netherlands.
How frequent is transient leukemia in Down syndrome children?
The frequency of TL is estimated to be around 10% in DS children, although this was studied in a selected (hospitalized) population. Most investigators assume that the true frequency is higher, but recent data suggest that his may not be the case. In The Netherlands, children with DS are registered in different nation-wide registries (the Dutch Pediatric Surveillance Unit and the National Dutch Neonatal and Obstetrical Registry), which provide population-based data. After matching these databases, it was assessed that 322 children (95% confidence interval 303–341) with DS were born in 2003, resulting in a prevalence of 16 out of 10,000 live-born babies. To date, five children with DS who were born in 2002 and 2003 have been diagnosed with myeloid leukemia (by the Dutch Childhood Oncology Group, E.R. van Wering, personal communication, 2007). As ML in patients with DS typically occurs before the age of 5 years, with a median age of approximately 2 years, and the frequency for progression from TL to ML DS is well established at 20%, these data suggest that the true frequency of TL is in the 3% to 5% range.
This is further supported by a recent study from Pine and colleagues, who screened Guthrie cards from 590 infants with DS for mutations in the GATA1 gene and found a mutation in 3.8% of children. However, GATA1 mutations may have been missed in those patients with minor preleukemic clones, subclonal mutations, low numbers of cells on Guthrie cards, or extramedullary TL without circulating blasts. In addition, one has to consider that some fetuses with DS may die in utero from causes that may include TL. Prospective population-based studies regarding the incidence of TL are currently underway in The Netherlands.
Down syndrome transient leukemia: clinical presentation
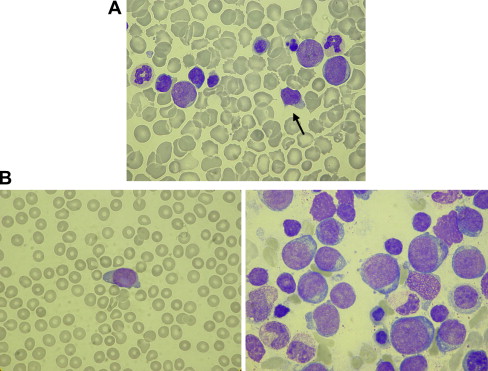
Massey and colleagues recently published the clinical data of a Children’s Oncology Group (COG) retrospective study in which 48 neonates with TL were enrolled. The median age at diagnosis of TL was 7 days (range 1–65 days). Approximately 25% of patients were a-symptomatic, although this study may have been biased toward registration of symptomatic patients. Hepatosplenomagaly (in 56% of cases), effusions (in 21%), and bleeding or petechiae (in 25%) were the most common findings in symptomatic infants. In terms of outcome, 9 out of 47 (19.1%) patients developed ML DS at a mean of 20 months (range 9–38 months). The development of subsequent leukemia was associated with cytogenetic abnormalities other than the constitutional +21, although there were no clear recurrent abnormalities. Eight out of 47 (17%) patients died early at a mean of 90 days. These eight children all had signs of liver failure and diffuse intravascular coagulation, as well as effusions (including pericardial, pleural, and ascitic effusions). Four patients had liver biopsies showing liver fibrosis, cholestasis, and infiltration with leukemic cells. None of the eight children had normalized their blood counts before death, although in three the blasts had disappeared completely from the peripheral blood. Overall, in 42 out of 47 (89%) patients the blasts disappeared after a mean of 58 days (range 2–194 days). Of these 42 children, four developed subsequent leukemia and three died early because of complications. The other 35 children did not have detectable hematologic abnormalities at follow-up, and normalized their blood counts at a mean of 84 days (range 2–201 days). See Fig. 2 A for an example of a blood smear of a TL patient.
The study from Massey and colleagues was recently confirmed by two other series, one from Japan and one from the AML-Berlin Frankfurt Münster Study Group (AML-BFM SG). The Japanese study evaluated 70 children with TL, and reported an early death rate of 23% and development of ML DS in 17% of surviving children. The AML-BFM SG registered 148 children with TMD between 1993 and 2006, mainly consisting of children with symptomatic TL (D. Reinhardt, unpublished data, 2007). Median white blood cell count at diagnosis was 40.3 × 10 9 per liter. Complete remission was achieved spontaneously in 66% of children; 19% of children were treated with low-dose cytarabine to relieve symptoms and achieved remission. Two children progressed to ML DS after 9 and 13 months of follow-up, without clearing their peripheral blood TL cells in the meantime. In total, 24 children died, including 21 in the first 23 months of life. Death was attributed to TL in 13 of these children, including seven cases with liver fibrosis. In most children (85%) blasts were cleared from the peripheral blood after 4 weeks. Of the 124 surviving children, 29 developed ML DS (23.4%) after a median remission of 1.5 years (0.5–3.0 years).
One of the most relevant questions unanswered to date is whether the development of ML DS following TL can be inhibited by treating children for TL. The AML BFM SG and the Dutch COG are currently performing a study to prospectively investigate this.
Clinical features of Down syndrome leukemias
Myeloid Leukemia of Down Syndrome: Clinical Characteristics and Classification
According to the World Health Organization (WHO) classification, AML requires the presence of at least 20% blasts in the bone marrow or peripheral blood. However, the pediatric literature suggests that this 20% threshold does not apply to ML in patients with DS. Overt leukemia in these children is preceded in 20% to 60% of cases by an indolent prephase of myelodysplasia (MDS), characterized by thrombocytopenia and dysplastic changes in the bone marrow, often with accompanying marrow fibrosis. This MDS may last several months or even years before progressing to overt leukemia. In contrast to MDS in non-DS children, which requires stem-cell transplantation for cure, MDS in children with DS shows a highly favorable response to chemotherapy alone. Therefore, Hasle and colleagues suggested that all cases of MDS and overt myeloid leukemia in DS, children should be classified as one disease entity, and referred to as “myeloid leukemia of Down syndrome” or ML DS. As this is a unique disease, it should be classified separately from other cases of AML in the WHO-classification.
The presenting characteristics of DS patients with ML differ from those with non-DS AML. In general, patients with ML DS are younger than 5 years of age at diagnosis, have a low diagnostic white blood cell count, and do not show meningeal involvement. Roughly two-thirds of cases are classified as acute megakaryoblastic leukemia (FAB M7), with the other patients being classified as FAB M0, M2, and M6 (see Fig. 2 B). Cytogenetic abnormalities, such as t(8;21) or inv(16), which can frequently be found in the blasts of non-DS AML patients, typically do not occur in patients with DS who develop ML. In contrast, somatic mutations of GATA1 are specific for ML DS. Additional copies of chromosome 8 and 21 (in addition to the constitutional trisomy 21) are the most frequently occurring cytogenetic abnormalities, and are found in approximately 10% to 15% each. Cytogenetic findings associated with a high rate of relapse in non-DS AML, such as monosomy 7 and −5/5q- also occur in DS patients (together in approximately 10%–20% of cases), but do not seem to have a negative impact on prognosis, although numbers are small.
Myeloid Leukemia in Down Syndrome: Treatment Outcome
DS children with ML have a superior outcome when compared with children without DS who develop AML. This was first recognized by investigators from the Pediatric Oncology Group (POG), and the Nordic Society for Pediatric Hematology and Oncology (NOPHO). However, to obtain excellent outcome rates it is crucial that high intensity treatment, which is required for cure of non-DS AML, is avoided, as this results in unacceptable high induction and treatment-related mortality rates in children with DS. This observation has led to specific treatment guidelines for children with DS who develop ML, mostly with reduced treatment intensity regimens. Recent results from several collaborative groups confirm the favorable outcome for this patient population. The 10-year overall survival (OS) for 61 patients treated according to the NOPHO between 1984 and 2001 was 74%. Similarly, the 5-year OS for 161 patients treated according to the Children’s Cancer Group study CCG 2891 between 1989 and 1999 was 79%. Finally, the 3-year OS for 67 children treated between 1998 and 2003, according to the AML-BFM 98 study, was 91%. Together, these data confirm a fairly favorable outcome for DS patients who develop myeloid leukemia.
Based on the excellent results in the AML-BFM 98 study, a prospective European treatment protocol was recently opened. DS patients with ML will receive the standard BFM chemotherapy regimen, with significant dose-reductions for cytarabine (28 g/m 2 versus 41 g/m 2 –47 g/m 2 for non-DS patients) and anthracyclines (230 mg/m 2 versus 320 mg/m 2 –450 mg/m 2 for non-DS patients). Moreover, DS patients will not receive maintenance therapy, nor cranial radiation, nor stem-cell transplantation. The COG has chosen a different approach in their 2971A trial, in which several drugs (etoposide and dexamethasone), as well as the 3-month maintenance period, were eliminated from the standard chemotherapy schedule used in the 2891 AML trial. In contrast to the European approach, there are no dose-reductions of the remaining drugs in the COG study, and the cumulative anthracycline and cytarabine dosages will be 320 mg/m 2 and 44 g/m 2 , respectively. Preliminary analysis shows outcome data (3-year probability of OS 84%) which are comparable to the CCG 2891 standard timing arm.
The potential for reduced treatment intensity is based on the unique hypersensitivity of myeloid leukemia cells of DS patients to chemotherapy, when compared with AML cells from non-DS individuals, as demonstrated by various investigators. This increased sensitivity to chemotherapy extends to agents with different mechanisms of action. Zwaan and colleagues have reported that myeloid blasts were significantly more sensitive to cytarabine (median, 12-fold), anthracyclines (2- to 7-fold), mitoxantrone (9-fold), amsacrine (16-fold), etoposide (20-fold), 6-thioguanine (3-fold), busulfan (5-fold) and vincristine (23-fold), than non-DS AML cells. Taub and colleagues have provided evidence for specific mechanisms to explain the observed differences in drug sensitivity, and linked sensitivity to a gene-dosage effect of chromosome 21 localized genes. For instance, cytarabine sensitivity may be caused by increased expression levels of the cystathionine-beta-synthase gene, which is located on chromosome 21. Other studies from this group have shown the impact of GATA1s on cytidine deaminase levels and cytarabine sensitivity. However, the fact that enhanced in-vitro sensitivity was described for drugs that exert therapeutic effect by different mechanisms of action may suggest a more general mechanism of enhanced chemotherapy susceptibility, such as a propensity to undergo apoptosis. Interestingly, ALL cells from DS patients do not differ in drug sensitivity when compared with those of patients without constitutional trisomy 21. This further supports the notion that ML in DS is a unique disease, and that sensitivity is cell-lineage specific rather than associated with a gene-dosage effect of genes located on chromosome 21.
It is currently unknown to what extent treatment intensity can be reduced in DS children with ML. In a small case series, a 77% overall survival in a selected group of patients (n = 18) was reported with a regimen consisting of cytarabine 10 mg/m 2 per dose subcutaneously, twice daily for 7 days every 2 weeks; vincristine 1 mg/m 2 intravenously every 2 weeks; and retinylpalmitate 250,000 units/m 2 per day orally. The NOPHO reported that approximately half of the patients included in their AML-93 protocol had dose-reductions of 75% and 67% of anthracyclines and cytarabine, respectively, which did not influence the relapse rate in those patients. These data suggest that further treatment reduction may be possible, at least in some patients. However, most studies have also reported resistant or relapsed cases of ML DS. Therefore, it would be helpful if investigators could identify subgroups of patients with ML DS, at low risk of resistant or relapsed disease, for whom further dose-reduction of treatment intensity is feasible.
So far, such prognostic subgroups have not been described, and most published series lack the statistical power to identify prognostic factors because of small patient numbers. Gamis and colleagues reported that children older than 4 years of age with ML DS had a poorer outcome than younger children. The older children, however, may have suffered from non-DS AML rather than typical DS associated ML, which occurs almost invariably in children younger than 5 years of age. Indeed, Hasle and colleagues have shown that only 2 out of 12 DS children over 4 years of age, with myeloid leukemia, had a GATA1 mutation. Hence, the authors suggest that ML occurring in children with DS may be classified based on GATA1 mutations as its unique molecular genetic basis. Cases lacking this mutation may benefit from treatment according to a protocol developed for non-DS AML, rather than treatment according to the less dose-intensive protocols developed for GATA1-mutation positive ML DS. It is still unclear whether real-time quantitative polymerase chain reaction (PCR) analysis for mutated GATA1 will be able to successfully measure minimal residual disease (MRD) levels, and therefore could be used for further risk stratification of ML in DS patients. The disadvantage of this assay is that it is laborious, given that GATA1 mutations are private. Alternative methods for MRD-detection that could be employed include flowcytometry or reverse transcription-PCR for the Wilms tumor gene WT1.
Acute Lymphoblastic Leukemia in Patients with Down Syndrome
In contrast to children with ML DS, children with ALL do not have a superior outcome when compared with their non-DS counterparts. Whitlock and colleagues, summarizing the CCG data on ALL in patients with DS treated between 1983 and 1995, reported that DS patients stratified to the standard risk ALL arm had a poorer outcome than those who did not have DS, while this difference was not present for those treated according to the high-risk ALL arm. This suggests that DS children with ALL may be under-treated when stratified in a standard risk arm, based on classical National Cancer Institute risk criteria (age 1–9 years and white blood cell count less than or equal to 50 ×10 9 /l). This was confirmed in the CCG 1952 study, which enrolled patients between 1996 and 2000. Another possible explanation for this observation could be that physicians may have dose-reduced DS children with ALL in fear of enhanced toxicity (see later in this article). This may also have contributed to the observed higher relapse rate in these reports, although details on dose intensity were not provided. In the BFM 2000 study, which used MRD-based stratification using T-cell receptor and immunoglobulin gene rearrangements, there were no significant differences between DS and non-DS ALL patients in MRD-based risk-group assignment, nor in day-15 marrow response or in antileukemic efficacy between DS and non-DS ALL. However, there was an increased rate of toxic deaths in the DS patients. At 5-years of follow-up, probability for event free survival was equivalent with 82% (standard error 5%) for DS children and 82% (standard error 1%) for non-DS children.
Despite a similar distribution over MRD-subgroups in the BFM study, there are clear biologic differences between DS and non-DS ALL cases. When considering favorable prognostic factors, the frequency of hyperdiploid (more than 50 chromosomes) ALL is lower in DS patients. The prognostically favorable trisomies, such as trisomy of chromosome 4, 10, and 17, are considerably less frequent in DS patients with ALL. Several, but not all investigators report a lower frequency of TEL-AML1 rearranged ALL in children with DS. Considering the unfavorable prognostic factors, there are neither cases of infant ALL with DS, nor CD10-negative pro-B ALL cases, while the frequency of T-cell ALL is lower in children with DS. In addition, unfavorable cytogenetic abnormalities, such as t(1;19), the Philadelphia chromosome, or MLL-gene rearrangements occur less frequently in DS ALL, although this was not confirmed by all groups. It needs to be stressed however, that the definitions for favorable and unfavorable characteristics in all these studies were based on ALL in non-DS patients. It is unknown whether these abnormalities have the same impact in the context of constitutional trisomy 21.
Similar to ML treatment, an optimal balance between antileukemic efficacy and treatment-related toxicity needs to be established for children with DS and ALL, who lack the typical enhanced chemotherapy sensitivity that characterizes the myeloid blasts in DS patients. There are two studies describing in vitro cellular drug resistance profiles of ALL lymphoblasts in DS, and both studies did not report enhanced sensitivity of DS lymphoblasts when compared with non-DS lymphoblasts. However, the data in these studies were not corrected for the differences in genetic make-up of DS and non-DS ALL, and therefore need to be interpreted with caution. Dördelmann and colleagues reported a tendency for ALL to have a better initial steroid response in DS patients. However this was not confirmed in the BFM 2000 study, which showed a nonsignificant difference of 5.8% nonresponders in non-DS ALL, and 3.2% in DS ALL cases.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


