Acute leukemias are hematologic malignancies characterized by increased numbers of myeloid or lymphoid blasts. The term “acute,” historically referring to a rapid onset and promptly fatal outcome, now indicates the relatively undifferentiated nature of the leukemic cells. The World Health Organization (WHO) Classification of hematopoietic and lymphoid tissue tumors is the most widely accepted and universally applied classification of acute leukemias with diagnostic criteria of disease entities based on the integrated combination of clinical, morphologic, immunophenotypic, and genetic characteristics.
1 The WHO Classification divides acute leukemias into myeloid, lymphoid, and ambiguous lineage (mixed) phenotypes, depending on the origin of the blast cell.
1
The overall annual incidence of these disorders in the general population is about 4 per 100,000, with approximately 70% of them being acute myeloid leukemia (AML). AML accounts for about 15% of childhood leukemias and for approximately 80% to 90% of acute leukemias in adults, with the median age at diagnosis being about 70 years. Acute lymphoblastic leukemia (ALL) is primarily a childhood disease, with the peak incidence between the ages of 2 and 3 years. It diminishes in frequency until it reaches a nadir from about the ages of 25 to 50, after which it increases to achieve a second, but minor, peak at ages older than 80.
The etiology of most cases is unknown, but a few patients have had previous exposure to ionizing radiation, cytotoxic chemotherapeutic agents, or chemicals such as benzene. Several congenital diseases, such as Down syndrome, Bloom syndrome, and Turner syndrome, have an increased incidence of AML, as do certain types of bone marrow failure, such as Fanconi anemia, the Blackfan-Diamond syndrome, and rare individuals with familial RUNX1 and ETV6 mutations. Very rare familial forms of acute leukemia (e.g., RUNX1, ETV6, ANKRD26 and GATA2 gene mutations) have also been recognized. Patients with myelodysplastic and myeloproliferative disorders have varying, but elevated risks of developing AML. Heavy cigarette smoking also increases the incidence.
At the time of diagnosis, most patients with acute leukemia have nonspecific symptoms, such as fatigue, lethargy, and weight loss. Some complaints, such as dyspnea, angina, and dizziness, arise from anemia. Fever from the disease itself or from an infection related to neutropenia can be the presenting manifestation. Bleeding, such as epistaxis or cutaneous ecchymoses, may occur from thrombocytopenia or from disseminated intravascular coagulation (DIC) in patients with acute promyelocytic leukemia (APL). Bone pain and tenderness can develop from bone marrow expansion or direct periosteal involvement. Gums may swell from leukemic infiltration, especially in the monocytic types of AML.
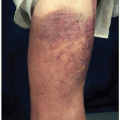
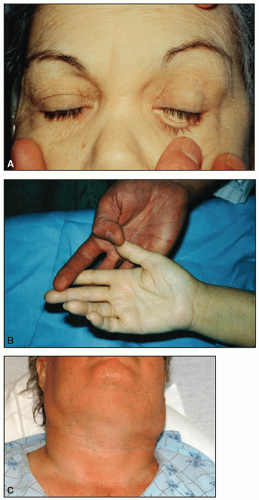
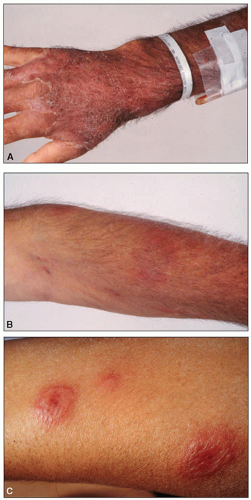
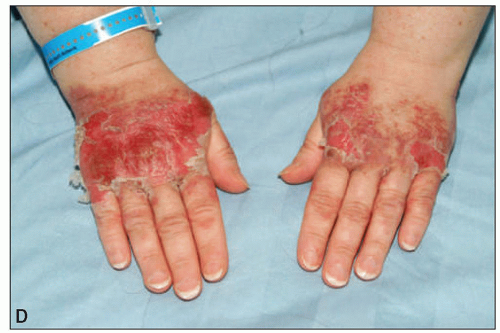
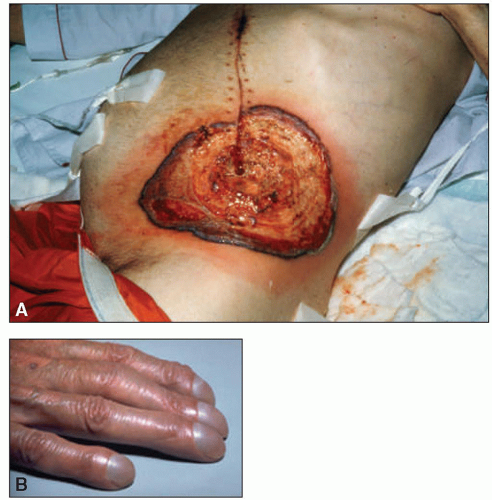
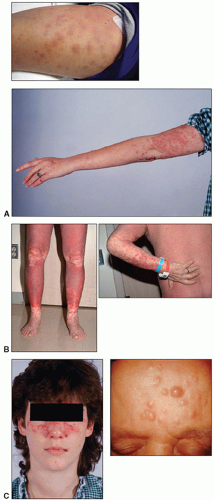
On physical examination, lymph node enlargement and hepatosplenomegaly may be detectable, more commonly in ALL than in AML. Pallor from anemia and petechiae or ecchymoses from thrombocytopenia or DIC may be apparent. Approximately 5% to 20% of patients with AML and ALL have skin infiltration with leukemic cells (leukemia cutis) sometime during the course of their disease. In about 5% to 10% of these patients, the cutaneous lesions precede the diagnosis, in about 35% to 45% they are simultaneous, and in about 55% they appear afterward, usually months later. They are typically erythematous or violaceous papules or nodules, but plaques, macules, palpable purpura, or ulcers also can occur. Patients with AML also may develop Sweet syndrome, characterized by an acute onset of fever and tender violaceous plaques that are often extensive, may affect the mucous membranes, and sometimes develop into blisters. Biopsies demonstrate mature neutrophils in the dermis. The fever and skin lesions disappear with systemic corticosteroid therapy, but recurrences are common with AML. Occasionally, prior to, or concurrent with, the diagnosis of AML, patients develop myeloid sarcomas, which are tumors of leukemic cells outside the bone marrow. These tumors can involve lymph nodes, skin, periosteum, extramedullary bone, and soft tissues. They often affect the subperiosteal bony structures of the skull, sternum, ribs, vertebrae, and pelvis.
At the time of presentation, the blood smear typically reveals decreased red cells and platelets, with the white count varying from leukopenia to marked leukocytosis. A decrease in the number of mature neutrophils is common. Circulating blasts are usually detectable. Aleukemic leukemia, in which blasts are not apparent in the peripheral blood, is slightly more common in AML than in ALL. With DIC, which occurs primarily with APL, microangiopathic features may be present, including red cell fragments, marked polychromatophilia, microcytes, and profound thrombocytopenia. In patients with preceding myelodysplastic syndromes (MDS), features of MDS are typically apparent, such as hypolobulated and hypogranular neutrophils, giant and agranular platelets, and erythrocytic macrocytosis and poikilocytosis.
All patients with suspected leukemia should undergo bone marrow aspirate and biopsy, with cytogenetic analysis, molecular testing, cytochemical analysis, and immunophenotyping done on the cells to delineate the correct classification of the leukemia. Usually, the presence of leukemia is obvious on bone marrow examination of Wright-Giemsa-stained aspirates: typically hypercellular, with sheets of blasts replacing the normally maturing cells in the erythroid, myeloid, and megakaryocytic lines. The distinction between a myeloid or lymphoid origin of blasts is crucial to classifying acute leukemia, as different types of acute leukemia require different treatment strategies. The presence in the cytoplasm of Auer rods—red, needle-like structures thought to be coalescences of primary granules—indicates a myeloblast. Otherwise, the distinction requires cytochemical or immunophenotypic studies. The first step is to use stains specific for myeloperoxidase (MPO) activity or for myeloid granules, and those that detect cells of monocytic lineage, such as nonspecific esterase. Concurrently, flow cytology is used to distinguish between minimally differentiated AML and ALL, to detect monocytic, erythroid or megakaryoblastic leukemia, to discriminate between B- and T-cell forms of ALL, and to establish the tumor-specific immunophenotypic signature that will be used in subsequent marrow samples in order to rule out minimal residual disease (MRD).
The criterion for the diagnosis of AML in most cases is that myeloblasts constitute at least 20% of the nucleated cells in the blood (based on counting 200 cells) or the bone marrow (counting 500 cells). The abnormal promyelocytes in APL and the promonocytes in AML with monocytic differentiation are considered blast equivalents. The diagnosis of AML can also be established in cases with evidence of t(8;21)(q22;q22) translocation, inv(16)(p13.1;q22) or t(15;17)(q22;q12) translocation regardless of the number of blasts or blast equivalents.
ACUTE MYELOID LEUKEMIAS
These disorders are defined as clonal expansions of myeloid blasts, most commonly in the blood or bone marrow, but occasionally presenting as tumor masses (myeloid sarcoma) in other tissues, such as skin and lymph nodes. The WHO classification (
Table 6.1) separates AML into seven general groups: (1) AML with recurrent genetic abnormalities; (2) AML with myelodysplasia-related changes; (3) therapy-related AML and MDS; (4) AML not otherwise categorized; (5) myeloid sarcoma; (6) myeloid proliferations related to Down syndrome; and (7) blastic plasmacytoid dendritic cell neoplasm. The last two entities are exceedingly rare in routine daily practice. The fourth group—AML not otherwise categorized—is a revision of the previously used FAB classification and divides cases of AML based on morphology and immunophenotype.
AML with Recurrent Genetic Abnormalities
These disorders have cytogenetic abnormalities, most commonly breaks in chromosomes in which the fragments join other chromosomes (translocations). These rearrangements create fusion genes that regulate the production of abnormal proteins.
AML with t(8;21)(q22;q22) (RUNX1-RUNXT1, previously called AML1-ETO) constitutes about 5% to 10% of AML cases, predominantly in younger patients. The
t(8;21)(q22;q22) translocation results in a fusion protein, RUNX1-RUNX1T1 (also called AML1-ETO) a transcription factor.
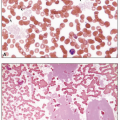
2 The blasts are typically large, with abundant basophilic cytoplasm, often with Auer rods and numerous, sometimes very large, azurophilic granules. Dysplasia in the form of abnormal nuclear segmentation and cytoplasmic staining may be present in promyelocytes, myelocytes, and mature neutrophils.
AML with inv(16)(p13q22) or t(16;16)(p13;q22) is found in about 10% of cases of AML, primarily in younger patients. The translocation results in a fusion protein, CBFB-MYH-11, a transcription factor.
3 The bone marrow usually has elements of both granulocytic (including myeloblasts) and monocytic differentiation (including monoblasts, promonocytes, and monocytes), combined with abnormal eosinophils. Eosinophil precursors contain abnormally large, purple granules that can be sufficiently numerous to obscure the nuclei.
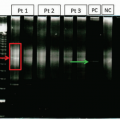
In addition to detection by FISH, the
RUNX1-RUNX1T1 and
CBFB-MYH11 transcript levels can be measured using quantitative RT-PCR (
Figure 6.24). The transcript levels can be used as a way to measure MRD, and serial monitoring of the transcript levels can be utilized to risk stratify and identify patients at high risk of relapse post-chemotherapy.
4AML with t(15;17)(q22;q12) or APL constitutes about 5% of AML. Abnormal promyelocytes are present, either hypergranular or hypogranular (microgranular). The
t(15;17)(q22;q12) translocation results in a fusion protein, PML-RARa, which leads to transcriptional repression and arrest of promyelocyte differentiation.
5 In the hypergranular form, the cytoplasm is packed with pink, red, or purple granules that are usually large, but may be fine. Bundles of Auer rods are present in most cases. The nuclei, which may be bilobed, are irregular in size and variable in shape, and may be reniform (kidney-shaped). APL is often associated with DIC, and has a high rate of early mortality due to hemorrhage if not
treated promptly. Treatment should be started immediately if morphology is suggestive, without waiting for confirmation by flow cytometry or cytogenetics.
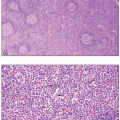
AML with 11q23 abnormalities, which constitutes about 5% of AML, occurs at any age, but is more common in children. Some cases develop after treatment with topoisomerase II inhibitors. Monocytic differentiation, with monoblasts and promonocytes predominating, is the most common morphologic pattern. Patients may have gum infiltration, leukemia cutis, and DIC. Monoblasts are large cells with round nuclei that usually contain lacy chromatin and large prominent nucleoli. The abundant basophilic and sometimes vacuolated cytoplasm may form pseudopods and contain scattered, fine azurophilic granules.
AML with Myelodysplasia-Related Changes
This type of AML occurring primarily in older adults has dysplasia in at least 50% of cells in two or more cell lines, including megakaryocytes. It is also diagnosed in patients with a previous diagnosis of MDS or MDS/MPN or in AML cases with an MDS-related cytogenetic abnormality. Abnormalities in granulopoiesis include hypogranular cytoplasm and hypolobulated or bizarrely segmented nuclei. Abnormal erythropoiesis is characterized by ring sideroblasts, vacuolated cytoplasm, and nuclei that are multiple, fragmented, or megaloblastic. Abnormal megakaryocytes are small or have single-lobed or multiple, discrete nuclei.
Therapy-Related AML
These occur as a consequence of cytotoxic drugs, radiation therapy, or both. One type follows alkylating agents or radiation therapy, most commonly about 5 years later. Myelodysplasia (MDS) usually occurs first, with evidence of bone marrow failure, to which many succumb without developing AML. About two-thirds of patients have MDS with multilineage dysplasia, and about 25% have refractory anemia with excess blasts. Whether during the MDS phase or after development into overt AML, these patients usually have dysplasia in all hematopoietic cell lines.
A second type of treatment-associated AML occurs after therapy with topoisomerase II inhibitors, such as etoposide and doxorubicin.
12 The average interval between the cytotoxic treatment and the occurrence of AML is about 33 months, usually without an intervening MDS phase. Most cases are acute myelomonocytic or monoblastic leukemias.
AML Not Otherwise Categorized
The classification depends on the morphologic and cytochemical characteristics of the blasts and their degree of differentiation and maturation.
AML, minimally differentiated, constitutes about 5% of AML and occurs mainly in adults. By morphology and light microscopic cytochemistry, the blasts show no myeloid differentiation. They are medium-sized, have an agranular basophilic cytoplasm, round or slightly indented nuclei with one or two nucleoli, and dispersed chromatin. On cytochemical studies fewer than 3% of the blasts react to Sudan black, α-naphthyl acetate, or stains that detect MPO.
AML, without maturation is responsible for about 10% of cases of AML, usually in adults. Azurophilic granules and Auer rods in the cytoplasm of the blasts may suggest their myeloid nature; in other cases, the blasts resemble lymphoblasts, from which they are differentiated by positivity to MPO stains or Sudan black in at least 3% of blast cells.
AML with maturation constitutes about 30% to 45% of cases of AML and may occur in all ages. Blasts may show azurophilic granules and Auer rods, and evidence of maturation is present with >10% of the marrow cells being promyelocytes, myelocytes, and mature neutrophils and <20% being monocytes. The neutrophils may show abnormally increased or decreased segmentation and lobulation. Basophils, eosinophils, and mast cells may be increased.
Acute myelomonocytic leukemia accounts for about 15% to 25% of AML, usually in the elderly and sometimes in patients who have had preceding chronic myelomonocytic leukemia. Both neutrophilic and monocytic cells and their precursors are present, each constituting at least 20% of the marrow cells. Circulating monocytes may be numerous (≥ 5 × 109/L). Monoblasts are large cells with round nuclei containing one or more prominent nucleoli and abundant basophilic cytoplasm, sometimes with fine azurophilic granules, vacuoles, and pseudopod formation. Promonocytes have a less basophilic and more granulated cytoplasm, containing occasional vacuoles and azurophilic granules. The nuclei are irregular and indented.
Acute monoblastic and acute monocytic leukemia each account for about 5% of AML, the former more common in children, the latter in adults. In both, at least 80% of the leukemic cells are in the monocytic line. In acute monoblastic leukemia, at least 80% of the monocytic cells are monoblasts; in acute monocytic leukemia, most of them are promonocytes.
Acute erythroid leukemias include two subtypes, erythroleukemia (erythroid/myeloid) and pure erythroid leukemia. The former constitutes about 5% of AML; the latter is very rare. In erythroleukemia, at least 50% of the nucleated cells in the bone marrow are erythroid and at least 20% of the nonerythroid cells are myeloblasts. The erythroid cells are dysplastic, containing multiple and megaloblastoid nuclei, the cytoplasm often possessing poorly delineated, coalescing vacuoles. The myeloblasts are similar to those in AML with and without maturation. Some cases of erythroleukemia evolve from a MDS. In pure erythroid leukemia, >80% of the marrow cells are erythroid. The erythroblasts have deeply basophilic, often agranular, cytoplasm that may contain poorly delineated vacuoles. The round nuclei have fine chromatin and one or more nucleoli.
Acute megakaryoblastic leukemia affecting all ages accounts for about 5% of AML. At least 50% of the blasts are from the megakaryocyte lineage. The megakaryoblasts are often pleomorphic and have a basophilic, often agranular, cytoplasm that may demonstrate pseudopod and bleb formation, indicating budding platelets. The nuclei have fine chromatin
and one to three nucleoli. Dysplastic platelets may be visible in the blood, as may be circulating micromegakaryocytes and megakaryocyte fragments.
Myeloid Sarcoma
AML can also manifest in extramedullary tissues, with or without bone marrow involvement. Myeloid sarcoma can occur as an isolated case, or more often occur in patients with history of leukemia or other myeloid malignancies.
6 Myeloid sarcoma can occur in the soft tissue, lymph nodes, or the periosteum.
7 Similarly, leukemia cutis can occur in the skin, most commonly extremities, back trunk, and face.
8 Extramedullary leukemia is diagnosed by obtaining biopsy of the affected site, and immunohistochemistry and flow cytometry are used to establish the diagnosis. Similar cytogenetic abnormalities associated with AML have been reported in these cases, and treatment is similar to that of AML, with systemic chemotherapy.
Acute Leukemias of Ambiguous Lineage
In less than 4% of cases of leukemia, diagnostic tests currently available have several drawbacks: (1) they cannot determine whether the blasts have a myeloid or lymphoid origin (acute undifferentiated leukemia); (2) they indicate two populations of cells, each having a distinct lineage from myeloid or T or B lymphocytes (acute bilineal leukemia); or (3) they indicate that the blasts individually have markers of two or three lines of myeloid, T lymphocytes, and B lymphocytes (acute biphenotypic leukemia). In acute undifferentiated leukemia, the blasts lack any distinguishing characteristics, whereas in the bilineal and biphenotypic forms, the leukemic cells may resemble lymphoblasts, myeloblasts, or monoblasts.


 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access