ACTIVATION OF THE MYOMETRIUM—PHASE 1 OF PARTURITION
Activation of the myometrium (phase 1 of parturition) can be considered as an active process involving the fetal genome. This influence may be exerted through a growth pathway that predisposes to uterine stretch and activation, or through an endocrine pathway (involving the fetal hypothalamic–pituitary–adrenal, or HPA, axis and increased output of cortisol), or through a combination of these processes. Phase 1 activation is manifested through up-regulation in the myometrium of a cassette of contraction-associated proteins (CAPs), including connexin-43 (CX-43; the major protein of gap junctions), and
receptors for oxytocin and stimulatory prostaglandins. Expression of increased gap junction protein allows formation of gap junctions, which permit cell-to-cell coupling, whereas expression of proteins that are constituents of ion channels determine the resting membrane potential and, hence, the excitability of the uterine myocytes.
receptors for oxytocin and stimulatory prostaglandins. Expression of increased gap junction protein allows formation of gap junctions, which permit cell-to-cell coupling, whereas expression of proteins that are constituents of ion channels determine the resting membrane potential and, hence, the excitability of the uterine myocytes.
EFFECTS OF STRETCH ON UTERINE ACTIVATION
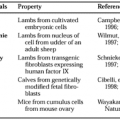
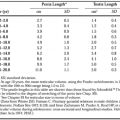
The effects of uterine stretch on CAP gene activation has been demonstrated in animal studies and is partly dependent on the prevailing endocrine environment. The normal prepartum increase in myometrial expression of CX-43, oxytocin receptor, and prostaglandin F receptor (FP) mRNA can be inhibited by administration of progesterone or induced by administration of the progesterone receptor antagonist mifepristone or by ovariectomy at earlier times in gestation. When a small (3-mm) inert tube was placed within one uterine horn of nonpregnant ovariectomized rats, a significant increase in CX-43 mRNA was seen compared with the contralateral horn, and this effect was blocked by progesterone. In rats made unilaterally pregnant, insertion of the inert tube into the nonpregnant horn increased expression of CX-43 and oxytocin receptor mRNA levels at the time of labor but was ineffective at earlier times of gestation when the endogenous, prevailing progesterone concentration was elevated.
Estrogen is also regarded as a uterotropin that promotes activation of myometrial function. In human pregnancy, estrogen production in the placenta depends on the provision of C-19 precursor steroids, predominantly from the fetal adrenal gland. The fetal zone of the fetal adrenal gland, which occupies ˜85% of the fetal adrenal cortex, is deficient in the enzyme 3β-HSD; therefore, it produces predominantly Δ5 steroids such as dehydroepiandrosterone (DHEA) that is secreted as its sulfoconjugate (DHEAS). Fetal DHEAS can be converted to estrone and estradiol in the placenta, and ˜50% of circulating maternal estrone and estradiol is derived from the placental aromatization of fetal DHEAS; the remainder is formed from maternal adrenal C-19 steroids. Activation of the pituitary-adrenal axis of the fetus occurs in late gestation. In subhuman primate species, the concentration of DHEAS in the fetal circulation shows a progressive increase at this time. This mirrors an increase in the maternal plasma estriol concentration (maternal estriol is formed in the placenta from the precursor 16α-hydroxy DHEAS, which is 90% of fetal origin and is formed in the fetal liver from adrenal DHEAS). The pattern of fetal adrenal activation reflected in DHEAS output resembles that seen in ruminant species, such as the sheep, in which a progressive increase in cortisol output occurs from the late-gestation fetal adrenal gland. In primates, the fetal adrenal is divided into the outer adult zone that produces predominantly aldosterone, the inner fetal zone that produces DHEAS, and a transitional zone, which is interposed between the adult and fetal cortex and is thought to produce cortisol. Adrenocorticotropic hormone (ACTH) stimulates steroidogenesis in the transitional and fetal zones. During pregnancy, fetal ACTH output may be relatively suppressed by the negative feedback of cortisol derived from the maternal circulation after transplacental transfer. In late gestation, increased activity of 11β-HSD (the type-2 isoform) in the placenta leads to increased conversion of maternal cortisol to cortisone, which is bioinactive. Therefore, less maternal cortisol enters the fetal compartment, and less negative feedback occurs, allowing fetal ACTH concentrations to rise.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


