DIFFERENTIAL DIAGNOSIS OF AN ABNORMAL SERUM PHOSPHORUS LEVEL
Frequently, a careful history and physical examination reveal the apparent cause of an abnormal serum phosphorus concentration. However, the variety of diseases, therapeutic agents, and physiologic states that affect phosphate homeostasis are numerous, and clinicians should make every effort to determine the mechanism underlying changes in the serum phosphorus
level in each patient. Determining the cause for the abnormality permits a rational choice of appropriate therapy.
level in each patient. Determining the cause for the abnormality permits a rational choice of appropriate therapy.
HYPERPHOSPHATEMIA
Two mechanisms generally underlie the genesis of hyperphosphatemia in humans. First and most commonly, reduced renal excretion of phosphate may cause phosphate retention. The reduction reflects either a reduced GFR or increased renal tubular reabsorption of phosphate (see Table 67-1). Second, hyperphosphatemia rarely ensues after an increase in phosphate intake or an acute increment of endogenous phosphate released into the extracellular fluid.
REDUCED RENAL PHOSPHATE EXCRETION
Renal Failure.
The most common cause of hyperphosphatemia is chronic renal failure. The increased serum phosphate concentration results from a decline of the GFR.11 However, in early stages of renal failure, compensatory changes in the renal tubular reabsorption of phosphate, in part because of elevated circulating levels of PTH, limit expression of this abnormality. Indeed, only at a GFR of ˜25 mL/min/l.73 m2 or less is hyperphosphatemia commonly seen. With advanced renal failure, however, serum phosphorus levels may increase up to a level of 11 or 12 mg/dL if dietary phosphate is high and phosphorus is freely mobilized from the skeleton by severe secondary hyperparathyroidism. Retention of phosphate in this disorder leads to the development of renal osteodystrophy, ectopic calcification (see later), depression of renal calcitriol synthesis, and impaired intestinal absorption of calcium. Hyperphosphatemia also lowers the serum ionized calcium and stimulates hypertrophy of the parathyroid glands and secretion of PTH.
Hypoparathyroidism.
Hyperphosphatemia characteristically complicates the hypoparathyroid states (idiopathic or postsurgical, or pseudohypoparathyroidism).12 The increased serum phosphate levels are secondary to enhanced renal reabsorption of phosphate. In idiopathic and postsurgical hypoparathyroidism, deficient PTH causes a loss of the hormonal effect to decrease the TmP. Conversely, the increased renal reabsorption of phosphate in the pseudohypoparathyroid disorders arises from renal resistance to PTH.
The concurrence of clinically significant hypocalcemia and hyperphosphatemia usually leads to the diagnosis of hypoparathyroidism. Some studies have revealed that the causes of idiopathic hypoparathyroidism in some affected patients include (a) gain-of-function calcium-receptor mutations that result in autosomal-dominant and sporadic disease13; (b) an autoimmune polyglandular syndrome caused by a genetic abnormality at 21q22.314; (c) an X-linked recessive inherited form of the disorder15; and (d) autoimmune-targeting of the calcium-sensing receptor.13 In contrast, investigations of the familial parathyroid hormone-resistance syndromes indicate that (a) pseudohypoparathyroidism type 1a occurs because of paternally imprinted mutations of GNAS1 on chromosome 20q13.11, resulting in abnormalities of theα subunit of the G protein, which couples receptor binding to activation of adenylate cyclase16; and (b) pseudohypoparathyroidism type 1b similarly manifests secondarily to a paternally imprinted mutation of the stimulatory G protein that maps to 20q13.3.17 In all of these disorders, treatment of the hypocalcemia with oral calcium supplements and vitamin D preparations may reduce but not necessarily normalize the serum phosphate levels and the renal TmP. This effect may be the result of the elevation of the serum calcium concentration, or perhaps it is the result of a direct effect of 1,25(OH)2D3 on renal tubular phosphate handling. Thus, patients with hypoparathyroidism who are successfully treated may have normal calcium and phosphate levels.
Tumoral Calcinosis.
Tumoral calcinosis is a rare genetic disease characterized by periarticular cystic and solid tumorous calcifications (Fig. 67-1). Specific biochemical markers of the
disorder include hyperphosphatemia and an elevated serum 1,25(OH)2D3 concentration. Using these criteria, evidence has been presented for autosomal-recessive inheritance of this syndrome. However, an abnormality of dentition, marked by short bulbous roots, pulp stones, and radicular dentin deposited in swirls, is a phenotypic marker of the disease that is variably expressed. Thus, this disorder may have multiple formes frustes that could complicate genetic analysis. Indeed, using the dental lesion as well as the more classic biochemical and clinical hallmarks of the disease, an autosomal-dominant pattern of transmission for tumoral calcinosis has been documented.18
disorder include hyperphosphatemia and an elevated serum 1,25(OH)2D3 concentration. Using these criteria, evidence has been presented for autosomal-recessive inheritance of this syndrome. However, an abnormality of dentition, marked by short bulbous roots, pulp stones, and radicular dentin deposited in swirls, is a phenotypic marker of the disease that is variably expressed. Thus, this disorder may have multiple formes frustes that could complicate genetic analysis. Indeed, using the dental lesion as well as the more classic biochemical and clinical hallmarks of the disease, an autosomal-dominant pattern of transmission for tumoral calcinosis has been documented.18
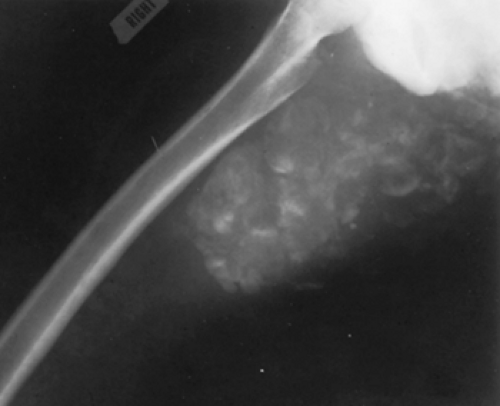 FIGURE 67-1. Tumoral calcinosis. Radiograph of right femur and pelvis of a 9-year-old boy with hyperphosphatemic tumoral calcinosis. This tumor was his fourth, and it had to be removed surgically. Serum phosphate level was 7.92 mg/dL (normal, 4.5–6.5 mg/dL); 1,25 dihydroxyvitamin D was 80 pg/mL (normal, 19–50 pg/mL). |
An increase in capacity of renal tubular phosphate reabsorption causes the hyperphosphatemia that is typical in affected subjects. Hypocalcemia is not a consequence of this abnormality, however, and the serum PTH concentration is normal. Moreover, the phosphaturic and urinary cyclic adenosine monophosphate (cAMP) responses to PTH are not disturbed. Thus, the defect does not represent renal insensitivity to PTH, or hypoparathyroidism. Rather, the basis of the disease is probably a primary abnormality of the renal tubule that enhances phosphate reabsorption. Undoubtedly, the calcific tumors result from the elevated calcium-phosphorus product. The observation that long-term phosphorus depletion alone or in association with administration of acetazolamide, a phosphaturic agent, leads to resolution of the tumor masses supports this possibility.19,20
An acquired form of this disease is rarely seen in patients with end-stage renal failure. Affected patients manifest hyperphosphatemia in association with either (a) an inappropriately elevated calcitriol level for the degree of renal failure, hyperparathyroidism, or hyperphosphatemia21,22; or (b) long-term treatment with calcium carbonate, calcitriol, or high calcium-content dialysates. Calcific tumors again likely result from an elevated calcium–phosphorus product. Indeed, complete remission of the tumors occurs on treatment with low-calcium dialysate23 or with vinpocetine, a mineral scavenger drug.24
Hyperthyroidism.
Up to one-third of patients with thyrotoxicosis may have hyperphosphatemia. The elevation of the serum phosphorus is caused by increased bone resorption and calcium mobilization, consequent PTH suppression, and an increase in renal tubular reabsorption of phosphate. The frequent concurrence of an elevated serum osteocalcin and alka-line phosphatase activity, as well as increased urinary calcium and hydroxyproline, indicates that thyroid hormone effects on bone cause the hyperphosphatemia.25,26 The increased TmP has been confirmed by direct measurements and by reports of elevated tubular reabsorption of phosphate in hyperthyroidism that reverts to normal after successful treatment. Secondary hypoparathyroidism, which is a response to thyroid hormone– associated hypercalcemia (see Chap. 42), is probably not the only explanation for the abnormal phosphate transport. Several studies suggest that thyroxine and triiodothyronine directly influence renal tubular phosphate reabsorption, but both enhanced and diminished phosphate transport have been reported. In any case, the hyperphosphatemia, when present, has no discernible effect on the clinical expression of the signs and symptoms of thyrotoxicosis.
Acromegaly.
Approximately two-thirds of patients with acromegaly have an elevated serum phosphate concentration that is secondary to the chronic effects of growth hormone on renal tubular reabsorption of phosphate27 (see Chap. 206). The serum phosphate level in affected subjects seldom exceeds 5.5 mg/dL but does correlate with disease activity. The mechanism underlying the increased phosphate reabsorption remains unknown. Although the administration of growth hormone to laboratory animals yields increased renal tubular phosphate reabsorption independent of alterations in the PTH concentration,28 somatotropin does not influence renal phosphate transport in a variety of in vitro systems. The widely distributed presence of insulin-like growth factor-I receptors in the kidney29 suggests that insulin-like growth factor-I probably mediates the effect of growth hormone on renal TmP (see Chap. 12).
Bisphosphonate Therapy.
The administration of disodium etidronate, a pyrophosphate analog, in doses generally higher than 5 mg/kg per day, can induce an increase in the serum phosphate concentration.30 The drug, which is used in the treatment of Paget disease and osteoporosis, causes a time-dependent and dose-dependent increment of the renal TmP and consequent phosphate retention that is largely responsible for the hyperphosphatemia. However, an inhibition of the normal intracellular translocation of phosphorus also contributes to the elevated serum levels. Although an elevation in serum phosphate levels occurs soon after the initiation of therapy, the maximum elevation of the phosphate concentration does not occur until after several days of treatment. The mechanism by which disodium etidronate alters the renal tubular reabsorption of phosphate is unclear. Drug therapy does not alter the serum PTH concentration. Moreover, the drug does not inhibit the urinary cAMP and phosphaturic response to infused PTH. A direct effect on renal phosphate transport, therefore, appears likely.
INCREASED PHOSPHATE LOAD
Vitamin D Intoxication.
An increase of the Pi load from exogenous sources generally does not cause hyperphosphatemia because the kidney excretes the excessive phosphorus. However, an increased serum phosphate concentration may occur in vitamin D intoxication when the gastrointestinal absorption of phosphate is markedly enhanced. Increased phosphate mobilization from bone and a reduction of GFR, secondary to hypercalcemia or nephrocalcinosis, may also contribute to the evolution of the hyperphosphatemia.
The chronic ingestion of large doses of vitamin D, in excess of 100,000 U per day, is generally required to cause intoxication. Suspected hypervitaminosis D may be investigated through competitive binding protein assays, which can document excessive amounts of vitamin D and its metabolites in the circulation (see Chap. 54 and Chap. 59).
Rhabdomyolysis.
Because muscle contains a large amount of phosphate, necrosis of muscle tissue may acutely increase the endogenous phosphate load and result in hyperphosphatemia. Such muscle necrosis (rhabdomyolysis) may complicate heatstroke, acute arterial occlusion, hyperosmolar nonketotic coma, trauma, toxic agents such as ethanol and heroin, and idiopathic paroxysmal myoglobinuria.31,32 Muscle biopsy often reveals myolytic denervation, and, as a consequence, acute renal failure caused by myoglobin excretion frequently complicates the clinical presentation and contributes to the hyperphosphatemia. However, an elevated serum phosphate concentration may precede evidence of renal failure or may occur in its complete absence when rhabdomyolysis is present. The diagnosis is confirmed by elevated serum creatine phosphokinase, uric acid, and lactate dehydrogenase concentrations and by the demonstration of heme-positive urine in the absence of red blood cells. Therapy is directed at the underlying disorder, with maintenance of the extracellular volume to avoid volume depletion and alkalinization of the urine to prevent uric acid accumulation and consequent acute tubular necrosis.
Cytotoxic Therapy.
Cytotoxic therapy often causes cell destruction and liberation of phosphorus into the circulation.33 The lysis of tumor cells begins within 1 to 2 days after treatment is initiated and is followed quickly by an elevation of the serum phosphate concentration. Hyperphosphatemia supervenes, however, only when the treated malignancies have a large tumor burden, rapid cell turnover, and substantial intracellular phosphorus content. Such malignancies include lymphoblastic leukemia, various types of lymphoma, and acute myeloproliferative syndromes, as well as solid tumors (e.g., small cell carcinoma, breast cancer, and neuroblastoma). Common risk factors for this syndrome include pretreatment renal insufficiency, elevated serum lactate dehydrogenase (LDH) concentration, and
hyperuricemia. Additional biochemical abnormalities observed in affected patients include hyperkalemia and hypocalcemia.
hyperuricemia. Additional biochemical abnormalities observed in affected patients include hyperkalemia and hypocalcemia.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


