Haemoglobin synthesis
Normal adult blood contains three types of haemoglobin (see Table 2.2). The major component is haemoglobin A with the molecular structure α2β2. The minor haemoglobins contain γ (fetal Hb or Hb F) or δ (Hb A2) globin chains instead of β chains. In the embryo and fetus, Gower 1, Portland, Gower 2 and fetal Hb dominate at different stages (Fig. 7.1). The genes for the globin chains occur in two clusters: ε, γ, δ and β on chromosome 11 and ζ and α on chromosome 16. Two types of γ chain occur, Gγ and Aγ, which differ by a glycine or alanine amino acid at position 136 in the polypeptide hypermethylchain. The α-chain gene is duplicated and both α genes (α1 and α2) on each chromosome are active (Fig. 7.1).
Figure 7.1 (a) The globin gene clusters on chromosomes 16 and 11. In embryonic, fetal and adult life different genes are activated or suppressed. The different globin chains are synthesized independently and then combine with each other to produce the different haemoglobins. The γ gene may have two sequences, which code for either a glutamic acid or alanine residue at position 136 (Gγ or Aγ, respectively). LCR, locus control region, HS-40, see text. (b) Synthesis of individual globin chains in prenatal and postnatal life.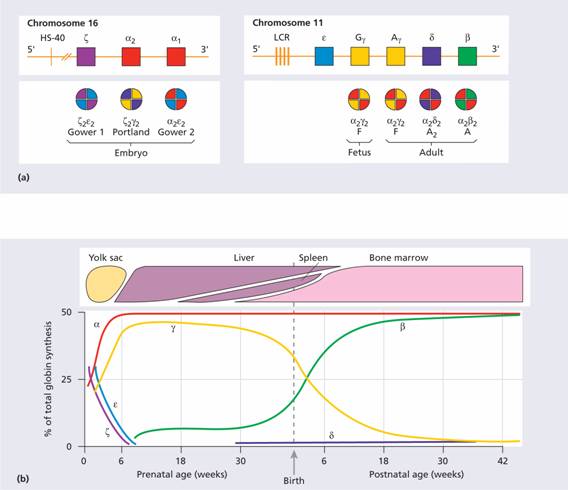
Molecular aspects
All the globin genes have three exons (coding regions) and two introns (non-coding regions whose DNA is not represented in the finished protein). The initial RNA is transcribed from both introns and exons, and from this transcript the RNA derived from introns is removed by a process known as splicing (Fig. 7.2). The introns always begin with a G-T dinucleotide and end with an A-G dinucleotide. The splicing machinery recognizes these sequences as well as neighbouring conserved sequences. The RNA in the nucleus is also ‘capped’ by addition of a structure at the 5’ end which contains a seven methyl-guanosine group. The cap structure may be important for attachment of the mRNA to ribosomes. The newly formed mRNA is also polyadenylated at the 3’ end (Fig. 7.2). This stabilizes it. Thalassaemia may arise from mutations or deletions of any of these sequences.
Figure 7.2 The expression of a humanglobin gene from transcription, excision of introns, splicing of exons and translation to ribosomes. The primary transcript is ‘capped’ at the 5’ end and a poly A tail is then added.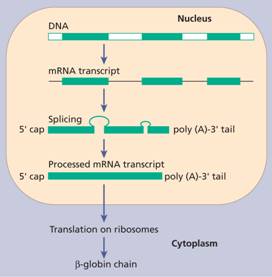
A number of other conserved sequences are important in globin synthesis and mutations at these sites may also give rise to thalassaemia. These sequences influence gene transcription, ensure its fidelity, specify sites for the initiation and termination of translation, and ensure the stability of newly synthesized mRNA. Promoters are found 5’ of the gene, either close to the initiation site or more distally. They are the sites where RNA polymerases bind and catalyse gene transcription (see Fig. 1.9). Enhancers occur either 5’ or 3’ to the gene (Fig. 7.2). Enhancers are important in the tissue-specific regulation of globin gene expression and in regulation of the synthesis of the various globin chains during fetal and postnatal life. The locus control region (LCR) is a genetic regulatory element, situated a long way upstream of the β-globin cluster, that controls the genetic activity of each domain, probably by physically interacting with the promoter region and opening up the chromatin to allow transcription factors to bind. The α-globin gene cluster also contains an LCR-like region termed HS40. GATA-1, FOG and NF-E2 transcription factors, expressed mainly in erythroid precursors, are important in determining the expression of globin genes in erythroid cells.
Globin mRNA enters the cytoplasm and attaches to ribosomes (translation) where the synthesis of globin chains takes place. This occurs by attachment of transfer RNAs, each with its individual amino acid, by codon–anticodon base pairing to an appropriate position on the mRNA template.
Switch from fetal to adult haemoglobin
The globin genes are arranged on chromosomes 11 and 16 in the order in which they are expressed (Fig. 7.1). Certain embryonic haemoglobins are usually only expressed in yolk sac erythroblasts. The β-globin gene is expressed at a low level in early fetal life, but the main switch to adult haemoglobin occurs 3–6 months after birth when synthesis of the γ chain is largely replaced by the β chain. BCL11A is a transcriptional regulator of the switch and of the silencing of δ chain synthesis in the adult. The methylation state of the gene (expressed genes tend to be hypomethylated, non-expressed hypermethylated), the state of the chromosome packaging and various enhancer sequences all play a part in determining whether a particular gene will be transcribed.
These result from the following:
1 Synthesis of an abnormal haemoglobin.
2 Reduced rate of synthesis of normal α- or β-globin chains (the α- and β-thalassaemias).
Table 7.1 shows some of the first group of syndromes that arise from synthesis of an α or β chain with an amino acid substitution. In many cases, however, the abnormality is completely silent. The clinically most important abnormality is sickle cell anaemia. Haemoglobin (Hb) C, D and E are also common and, like Hb S, are substitutions in the β chain. Unstable haemoglobins are rare and cause a chronic haemolytic anaemia of varying severity with intravascular haemolysis (see Table 6.2). Abnormal haemoglobins may also cause (familial) polycythaemia (see Chapter 15) or congenital methaemoglobinaemia (see Chapter 2).
Table 7.1 The clinical syndromes produced by haemoglobin abnormalities.
| Syndrome | Abnormality |
| Haemolysis | Crystalline haemoglobins (Hb S, C, D, E, etc.) Unstable haemoglobin |
| Thalassaemia | α or β resulting from reduced globin chain synthesis |
| Familial polycythaemia | Altered oxygen affinity |
| Methaemoglobinaemia | Failure of reduction (Hb Ms) |
The genetic defects of haemoglobin are the most common genetic disorders worldwide. They occur in tropical and subtropical areas (Fig. 7.3) and most appear to have been selected because the carrier state affords some protection against malaria.
Figure 7.3 The geographical distribution of the thalassaemias and the more common inherited structural haemoglobin abnormalities.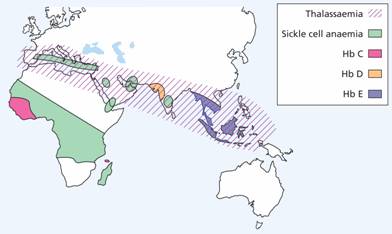
These are a heterogeneous group of genetic disorders that result from a reduced rate of synthesis of α or β chains. β-Thalassaemia is more common in the Mediterranean region while α-thalassaemia is more common in the Far East.
α-Thalassaemia syndromes
These are usually caused by gene deletions and are listed in Table 7.2. As there are normally four copies of the α-globin gene, the clinical severity can be classified according to the number of genes that are missing or inactive. Loss of all four genes completely suppresses α-chain synthesis (Fig. 7.4) and because the α chain is essential in fetal as well as in adult haemoglobin this is incompatible with life and leads to death in utero (hydrops fetalis; Fig. 7.5). Three α gene deletions leads to a moderately severe (haemoglobin 7–11 g/dL) microcytic, hypochromic anaemia (Fig. 7.6) with splenomegaly. This is known as Hb H disease because haemoglobin H (β4) can be detected in red cells of these patients by electrophoresis or in reticulocyte preparations (Fig. 7.6). In fetal life, Hb Barts (γ4) occurs.
Table 7.2 Classification of thalassaemia.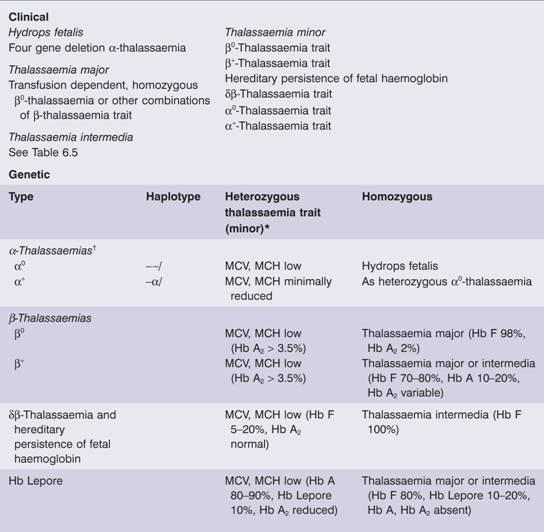
MCH, mean corpuscular haemoglobin; MCV, mean corpuscular volume.
*Occasionally heterozygous β – thalassaemia is dominant (associated with the clinical picture of thalassaemia intermedia).
There are several explanations.
†Compound heterozygote α0α+ (– –/–α) is haemoglobin H disease.
Figure 7.4 The genetics of α – thalassaemia. Each α gene may be deleted or (less frequently) dysfunctional. The orange boxes represent normal genes, and the blue boxes represent gene deletions or dysfunctional genes.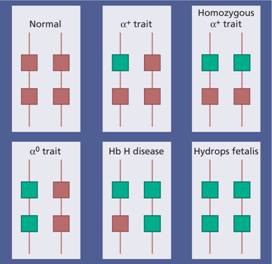
Figure 7.5 α-Thalassaemia: hydrops fetalis, the result of deletion of all four α-globin genes (homozygous α0-thalassaemia). The main haemoglobin present is Hb Barts (γ4). The condition is incompatible with life beyond the fetal stage. (Courtesy of Professor D. Todd)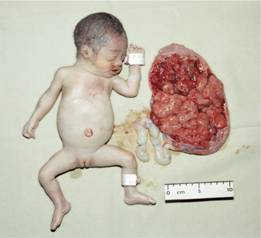
Figure 7.6 (a) α-Thalassaemia: haemoglobin H disease (three α-globin gene deletion). The blood film shows marked hypochromic microcytic cells with target cells and poikilocytosis. (b) α-Thalassaemia: haemoglobin H disease. Supravital staining with brilliant cresyl blue reveals multiple fine, deeply stained deposits (‘golf ball’ cells) caused by precipitation of aggregates of β-globin chains. Hb H can also be detected as a fast-moving band on haemoglobin electrophoresis (Fig. 7.12).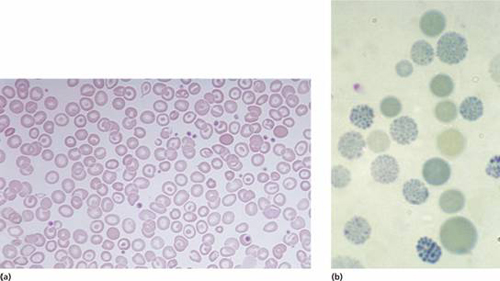
The α-thalassaemia traits are caused by loss of one or two genes and are usually not associated with anaemia, although the mean corpuscular volume (MCV) and mean corpuscular haemoglobin (MCH) are low and the red cell count is over 5.5 × 1012/L. Haemoglobin electrophoresis is normal and DNA analysis is needed to be certain of the diagnosis. Uncommon non-deletional forms of α-thalassaemia are caused by point mutations producing dysfunction of the genes or rarely by mutations affecting termination of translation which give rise to an elongated but unstable chain (e.g. Hb Constant Spring). Two rare forms of α-thalassaemia are associated with mental retardation. They are caused by mutation in a gene on chromosome 16 (ATR-16) or on chromosome X (ATR-X) which control the transcription of the α globin and other genes.
β-Thalassaemia syndromes
β-Thalassaemia major
This condition occurs on average in one in four offspring if both parents are carriers of the β-thalassaemia trait. Either no β chain (β0) or small amounts (β+) are synthesized. Excess α chains precipitate in erythroblasts and in mature red cells causing the severe ineffective erythropoiesis and haemolysis that are typical of this disease. The greater the α-chain excess, the more severe the anaemia. Production of γ chains helps to ‘mopup’ excess α chains and to ameliorate the condition. Over 400 different genetic defects have now been detected (Figs 7.7 and 7.8).
Figure 7.7 Distribution of different mutations of β-thalassaemia major in the Mediterranean area. IVSI, IVS2 intervening sequences; 1, 6, 39, 110, 745 are mutations of corresponding codons. (Courtesy of Professor A. Cao.)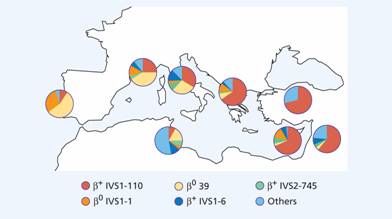
Figure 7.8 Examples of mutations that produce β-thalassaemia. These include single base changes, small deletions and insertions of one or two bases affecting introns, exons or the flanking regions of the β-globin gene. FS, ‘frameshifts’: deletion of nucleotide(s) that places the reading frame out of phase downstream of the lesion; NS, ‘non-sense’: premature chain termination as a result of a new translational stop codon (e.g. UAA); SPL, ‘splicing’: inactivation of splicing or new splice sites generated (aberrant splicing) in exons or introns; promoter, CAP, initiation: reduction of transcription or translation as a result of lesion in promoter, CAP or initiation regions; Poly A, mutations on the poly A addition signal resulting in failure of poly A addition and an unstable mRNA.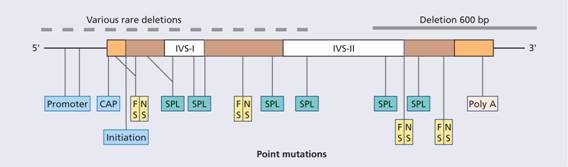
Unlike α-thalassaemia, the majority of genetic lesions are point mutations rather than gene deletions. These mutations may be within the gene complex itself or in promoter or enhancer regions. Certain mutations are particularly frequent in some communities (Fig. 7.7) and this may simplify antenatal diagnosis aimed at detecting the mutations in fetal DNA. Thalassaemia major is often a result of inheritance of two different mutations, each affecting β-globin synthesis (compound heterozygotes). In some cases, deletion of the β gene, δ and β genes or even δ, β and γ genes occurs. In others, unequal crossing-over has produced δβ fusion genes (so – called Lepore syndrome named after the first family in which this was diagnosed) (see p. 99).
Clinical features
1 Severe anaemia becomes apparent at 3–6 months after birth when the switch from γ- to β-chain production should take place.
2 Enlargement of the liver and spleen occurs as a result of excessive red cell destruction, extramedullary haemopoiesis and later because of iron overload. The large spleen increases blood requirements by increasing red cell destruction and pooling, and by causing expansion of the plasma volume.
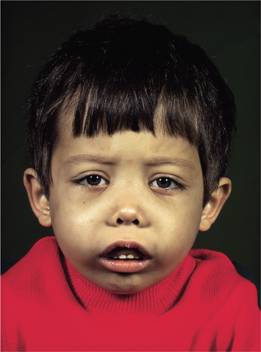
3 Expansion of bones caused by intense marrow hyperplasia leads to a thalassaemic facies (Fig. 7.9) and to thinning of the cortex of many bones with a tendency to fractures and bossing of the skull with a ‘hair-on-end’ appearance on X-ray (Fig. 7.10).
Figure 7.9 The facial appearance of a child with β-thalassaemia major. The skull is bossed with prominent frontal and parietal bones; the maxilla is enlarged.