Hereditary coagulation disorders
Hereditary deficiencies of each of the coagulation factors have been described. Haemophilia A (factor VIII deficiency), haemophilia B (Christmas disease, factor IX deficiency) and von Willebrand disease (VWD) are the most frequent; the others are rarer.
Haemophilia A is the most common of the hereditary clotting factor deficiencies. The prevalence is of the order of 30–100 per million population. The inheritance is sex-linked (Fig. 26.1) but up to one-third of patients have no family history and result from recent mutation.
Figure 26.1 A typical family tree in a family with haemophilia. Note the variable levels of factor VIII activity in carriers (*) because of random inactivation of X chromosome (Lyonization). The percentages show the degree of factor VIII activity as a percentage of normal.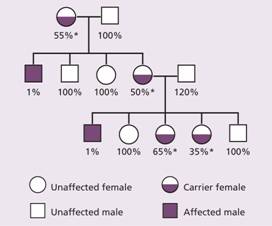
Molecular genetics
The factor VIII gene is situated near the tip of the long arm of the X chromosome (Xq2.8 region). It is extremely large and consists of 26 exons. The factor VIII protein includes a triplicated region A1A2A3 with 30% homology with each other, a duplicated homology region C1C2 and a heavily glycosylated B domain which is removed when factor VIII is activated by thrombin. The protein is synthesized in the liver and endothelial cells.
The defect is an absence or low level of plasma factor VIII. Approximately half of the patients have missense or frameshift mutations or deletions in the factor VIII gene. In others a characteristic ‘flip-tip’ inversion is seen in which the factor VIII gene is broken by an inversion at the end of the X chromosome (Fig. 26.2). This mutation leads to a severe clinical form of haemophilia A.
Figure 26.2 The mechanism of the flip-tip inversion leading to disruption of the factor VIII gene. (Left) The orientation of the factor VIII gene is shown with the three copies of gene A (F8A) in this region (one within an intron 22 and two near the telomere). (Middle) During spermatogenesis at meiosis, the single X pairs with the Y chromosome in the homologous regions. The X chromosome is longer than the Y and there is nothing to pair with most of the long arm of X. The chromosome undergoes homologous recombination between the A genes. (Right) The final result is that the factor VIII gene is disrupted. cen, centromeric end; tel, telomere; the arrows indicate the direction of transcription from the A gene.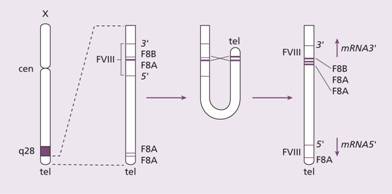
Clinical features
Infants may develop profuse post-circumcision haemorrhage or joint and soft tissue bleeds and excessive bruising when they start to be active. Recurrent painful haemarthroses and muscle haematomas dominate the clinical course of severely affected patients and if inadequately treated lead to progressive joint deformity and disability (Figs 26.3–26.6). Local pressure can cause entrapment neuropathy or ischaemic necrosis. Prolonged bleeding occurs after dental extractions. Spontaneous haematuria and gastrointestinal haemorrhage, sometimes with obstruction resulting from intramucosal bleeding, can also occur. The clinical severity of the disease correlates inversely with the factor VIII level (Table 26.1). Operative and post-traumatic haemorrhage are life-threatening both in severely and mildly affected patients. Although not common, spontaneous intracerebral haemorrhage occurs more frequently than in the general population and is an important cause of death in patients with severe disease.
Figure 26.3 Haemophilia A: acute haemarthrosis of the right knee joint with swelling of the suprapatellar region. There is wasting of the quadriceps muscles, particularly on the left.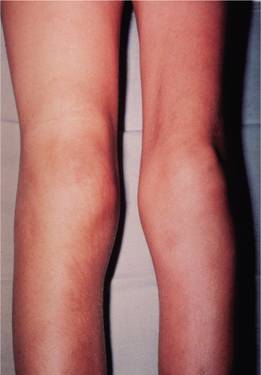
Figure 26.4 Haemophilia A showing severe disability. The left knee is swollen with posterior subluxation of the tibia on the femur. The ankles and feet show residual deformities of talipes equinus, with some cavus and associated toe clawing. There is generalized muscle wasting. The scar on the medial side of the left lower thigh is the site of a previously excised pseudotumour.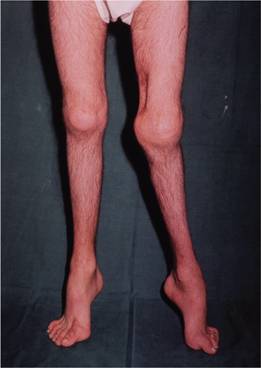
Figure 26.5 (a) Haemophilia A: massive haemorrhage in the area of the right buttock. (b) 15-year-old boy with sudden left hip pain and haemophilia A. Magnetic resonance imaging (MRI) axial image, T2 weighted, revealing large left spontaneous haematoma (yellow arrow) in left gluteus maximus muscle compared with normal right side (red cross). (Courtesy of Dr P. Wylie.)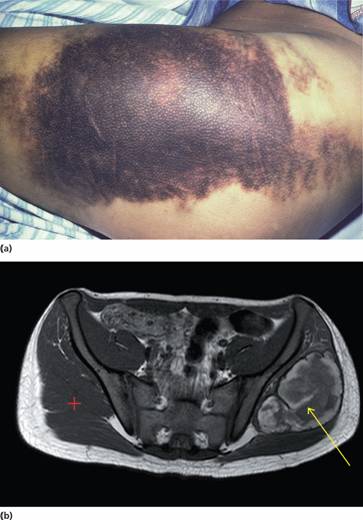
Figure 26.6 Haemophilia A: X-ray of the knee joints shows destruction and narrowing of the left joint space.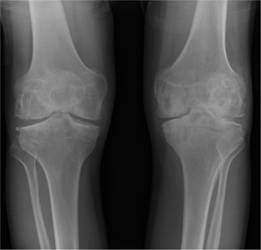
Table 26.1 Correlation of coagulation factor activity and disease severity in haemophilia A or B.
| Coagulation factor activity (percentage of normal) | Clinical manifestations |
| <1 | Severe disease |
| Frequent spontaneous bleeding into joints, muscles, internal organs from early life | |
| Joint deformity and crippling if not adequately prevented or treated | |
| 1–5 | Moderate disease |
| Bleeding after minor trauma | |
| Occasional spontaneous episodes | |
| >5 | Mild disease |
| Bleeding only after significant trauma, surgery |
Haemophilic pseudotumours are large encapsulated haematomas with progressive cystic swelling from repeated haemorrhage. They are best visualized by magnetic resonance imaging (MRI) (Fig. 26.5b). They may occur in fascial and muscle planes, large muscle groups and in the long bones, pelvis and cranium. The latter result from repeated subperiosteal haemorrhages with bone destruction and new bone formation.
As a result of human immunodeficiency virus (HIV) present in concentrates made from human plasma during the early 1980s, over 50% of haemophiliacs treated in the USA or Western Europe became infected with HIV. Acquired immune deficiency syndrome (AIDS) has been a common cause of death in severe haemophilia. Thrombocytopenia from HIV infection may exacerbate bleeding episodes.
Many patients were infected with hepatitis C virus before testing of donors and blood products became possible. This has resulted in chronic hepatitis, cirrhosis and hepatoma. Hepatitis B transmission may also be a risk. Liver transplantation cures the haemophilia.
Laboratory findings (Table 26.2)
The following tests are abnormal:
1 Activated partial thromboplastin time (APTT).
2 Factor VIII clotting assay.
The platelet function analysis-100 (PFA-100) (and bleeding time) and prothrombin time (PT) are normal.
Table 26.2 Main clinical and laboratory findings in haemophilia A, factor IX deficiency (haemophilia B, Christmas disease) and von Willebrand disease.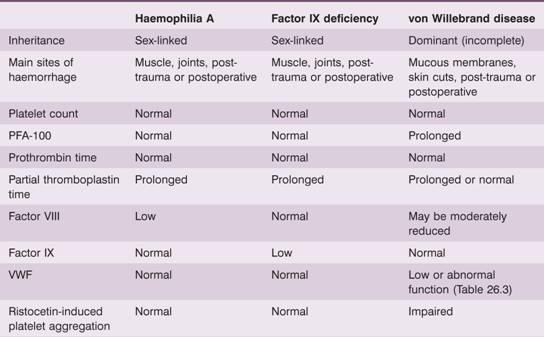
VWF, von Willebrand factor.
Carrier detection and antenatal diagnosis
Carriers are detected with DNA probes. A known specific mutation can be identified (Fig. 26.7) or restriction fragment length polymorphisms within or close to the factor VIII gene allow the mutant allele to be tracked. Chorionic biopsies at 8–10 weeks ’ gestation provide sufficient fetal DNA for analysis. Antenatal diagnosis is also possible following the demonstration of low levels of factor VIII in fetal blood obtained at 16–20 weeks ’ gestation from the umbilical vein by ultrasound-guided needle aspiration. This method is now only used if DNA analysis is uninformative (1% of carriers).
Figure 26.7 Family I: Haemophilia A: carrier detection by DNA analysis of peripheral blood leucocytes. (a) Normal reference sequence. (b) Automated dideoxy sequencing of the factor VIII gene has shown the affected son in his family (II:3) to have a 2 base pair (CT) deletion in exon 14, resulting in a frameshift causing introduction of a premature stop codon. (c) His mother is shown to be a carrier illustrated by one allele showing normal sequence and the second carrying the 2 base pair deletion. His older sister (II:2) was also a carrier, whereas his younger sister (II:4) was normal. (Courtesy of Haemophilia Centre, Royal Free Hospital, London.)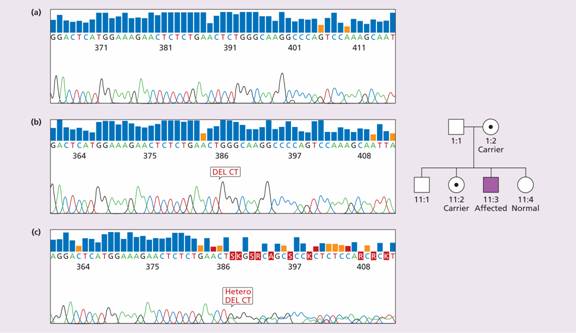
Treatment
Most patients in developed countries attend specialized haemophilia centres where there is a multidisciplinary team dedicated to their care. Bleeding episodes are treated with factor VIII replacement therapy and spontaneous bleeding is usually controlled if the patient ’ s factor VIII level is raised to 30–50% of normal. Guidelines exist for the plasma level to be achieved for different types of haemorrhage. For major surgery, serious post-traumatic bleeding or when haemorrhage is occurring at a dangerous site, the factor VIII level should be elevated to 100% and then maintained above 50% when acute bleeding has stopped, until healing has occurred. On average, factor VIII infusion produces a plasma increment of 2 U/dL per unit infused per kilogram body weight. Roughly, the dose to be infused (units) = (weight (kg) × increment needed (U/dL))/2.
Recombinant factor VIII and plasma-derived purified factor VIII preparations, which are heat and solvent-detergent treated, are available for clinical use and have never transmitted viral infections.
1-Diamino-8-D-arginine vasopressin (DDAVP; desmopressin) provides an alternative means of increasing the plasma factor VIII level in milder haemophiliacs. Following the intravenous administration of this drug, there is a two-to fourfold rise maximum at 30–60 min in the patient ’ s own factor VIII by release from endothelial cells and this rise is proportional to the resting level. DDAVP may also be taken subcutaneously or nasally–this has been used as immediate treatment for mild haemophilia after accidental trauma or haemorrhage. DDAVP has an antidiuretic action and should be avoided in the elderly; fluid restriction is advised after its use.
Local supportive measures used in treating haemarthroses and haematomas include resting the affected part, application of ice and the prevention of further trauma.
Prophylactic treatment
The increased availability of factor VIII concentrates that may be stored in domestic refrigerators has dramatically altered haemophilia treatment. At the earliest suggestion of bleeding, the haemophilic child may be treated at home. This advance has reduced the occurrence of crippling haemarthroses and the need for inpatient care. Severely affected patients are now reaching adult life with little or no arthritis. After the first spontaneous joint bleed, most boys with severe haemophilia are started on prophylactic factor VIII three times a week, aiming to keep their factor VIII trough levels above 1%. This may require the placement of a vascular access device such as Port-a-Cath if venous access is difficult. A controlled trial has proven that regular prophylaxis is far superior to on-demand treatment as judged by progression of joint damage, which was virtually absent in children on prophylaxis but always seen in boys treated on-demand.
Haemophiliacs are advised to have regular conservative dental care. Haemophiliac children and their parents often require extensive help with social and psychological matters. With modern treatment the lifestyle of a haemophilic child can be almost normal but certain activities such as body extreme contact sports are to be avoided, or undertaken with extra prophylaxis.
Gene therapy
Because it is only necessary to maintain factor levels > 1% to prevent most of the mortality and morbidity of factor VIII or IX deficiency, there is great interest in gene-based therapy. Various viral vectors (retroviral, adeno-associated) as well as non-viral vectors are being explored. Phase 1 trials are being carried out for both haemophilia A and B.
Inhibitors
One of the most serious complications of haemophilia is the development of antibodies (inhibitors) to infused factor VIII which occurs in 30–40% of severely effected patients, usually within the first 50 days of exposure. This renders the patient refractory to further replacement therapy. Immunosuppression and immune tolerance regimens have been used in an attempt to eradicate the antibody with success (at great cost) in about two-thirds of cases. Recombinant activated factor VII (VIIa) and activated prothrombin complex concentrates (FEIBA–factor VIII inhibitor bypassing activity) can be useful in the treatment of bleeding episodes.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


