Figure 37-1 Intra-abdominal spread of ovarian cancer. Ovarian cancers metastasize through lymphatics to lymph nodes at the level of the renal hilus, through blood vessels to the liver and other organs, but most frequently over the surface of the parietal and visceral peritoneum from pelvis to diaphragm. Blast Jr RC, Mills GB. Molecular pathogenesis of epithelial ovarian cancer. In: Mendelsohn J, Howley P, Israel M, Gray JW, Thompson CB, eds. The Molecular Basis of Cancer, 3rd ed. Philadelphia, Pa: Saunders-Elsevier; 2008:441-454.
Given the location of the ovaries within the pelvic cavity and the difficulty in assessing abnormalities on routine gynecologic examination, the disease is diagnosed only after it has metastasized in approximately 80% of cases. Ovarian cancer is often described as a “silent killer,” but the disease is generally symptomatic, even at early stages in 89% of cases. 8 Symptoms are not, however, specific and are generally attributed to benign gastrointestinal, genitourinary, musculoskeletal, or gynecologic conditions. Detection of a pelvic mass by physical examination or transvaginal sonography generally prompts exploratory surgery to remove the primary tumor and as much of the metastatic disease as possible—so-called cytoreductive surgery. Chemotherapy is generally given for 18 weeks thereafter using a combination of cytotoxic drugs including a taxane (paclitaxel or docetaxel) and a platinum derivative administered intravenously or directly into the peritoneal cavity.
In the 20% of patients with disease that is still localized to the ovaries (stage I), the prognosis is excellent, with up to 90% survival at 5 years using currently available surgery and chemotherapy. As the disease spreads to the other pelvic organs (stage II), to the peritoneal cavity and retroperitoneum (stage III), or to the hepatic parenchyma, pleural cavity, or lymph nodes outside the abdomen (stage IV), the prognosis becomes progressively worse, with a 5-year survival of less than 10% in the last group. Overall, 5-year survival rates have improved significantly (P < .05) from 37% in the 1970s to 45% in the 2000s, 2 related in large part to improvements in cytoreductive surgery and combination chemotherapy with carboplatin and paclitaxel. Over the past decade, however, median 5-year survival has not improved for patients with newly diagnosed advanced-stage ovarian cancer treated on clinical protocols of the Gynecologic Oncology Group. Moreover, long-term survival for women with advanced disease has not improved dramatically over the past three decades, and 70% of patients eventually succumb to the disease.
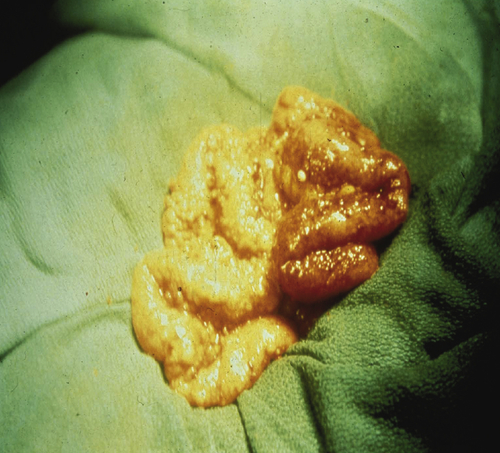
Figure 37-2 Intraperitoneal metastases from epithelial ovarian cancer studding the peritoneal surface.
Cellular and Molecular Characteristics of Ovarian Cancer Cells
Heterogeneity of Ovarian Cancers
As in many other malignancies, epithelial ovarian cancer is a clonal disease that arises from a single cell in more than 90% of cases. 9 Despite a clonal origin, epithelial ovarian cancers exhibit marked heterogeneity at a molecular, cellular, and clinical level.
Cell Proliferation
Among cancers from different patients with invasive cancer, the fraction of cycling cells can vary from 1% to 79% with a mean of 9% to 34% in different series. 10 Cyclin D1, cyclin E, and CDK2 are upregulated in a minority of cancers with their DNA copy numbers or protein levels correlating inversely with survival. Conversely, the p16, p21, and p27 CDK inhibitors are downregulated or mislocalized in a fraction of cancers, associated with a poorer outcome.
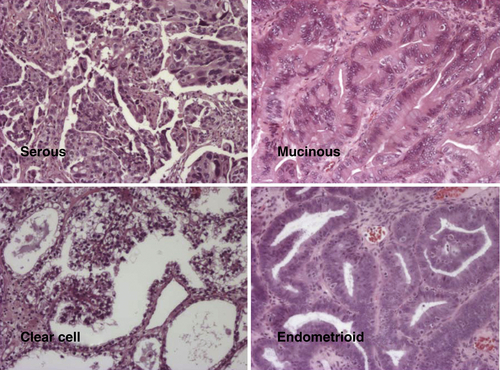
Figure 37-3 Different histotypes of epithelial ovarian cancer.
Histotypes
Ovarian cancers exhibit distinct histotypes—serous, endometrioid, clear cell, mucinous—that resemble epithelial components of normal fallopian tube, endometrium, vagina, endocervix, or intestine (Figure 37-3 ). Histotypes differ with regard to risk factors, genetic abnormalities, expression of tumor markers, and response to chemotherapy. 10 Each histotype exhibits a distinctive pattern of gene expression judged by array analysis, real-time reverse transcriptase–polymerase chain reaction (RT-PCR), and immunohistochemistry. 11 Molecular alterations in ovarian cancers of different histotypes correlate with changes in the normal tissues that they resemble morphologically. The HOX family of homeobox genes plays an important role in determining the histotype of ovarian cancers. 12 During normal development, HOXA9 is solely expressed in the primordia of the fallopian tubes, HOXA10 in the developing uterus, HOXA11 in the lower uterine segment and cervix, and HOXA13 in the future upper vagina. Expression of these HOX genes is retained in adult tissues but is not observed in ovarian surface epithelia. Expression of HOXA9, HOXA10, and HOXA11 is recapitulated in serous, endometrioid, and mucinous epithelial ovarian cancers, and enforced alterations in expression alter cellular histotype, indicating a causal role. 12
Low and High Grade (Type I and II)
Low- and high-grade ovarian cancers differ not only in histologic differentiation, but also in pathogenesis, genotype, rate of growth, prognosis, and response to therapy, permitting separation into Type I (low-grade) and Type II (high-grade) lesions. 13–15 Type I cancers include low-grade serous, mucinous, endometrioid, and clear cell histotypes and are often diagnosed in early stage (I or II), grow slowly, and resist conventional chemotherapy. Low-grade tumors frequently express estrogen receptors and may respond to tamoxifen or aromatase inhibitors. The more prevalent Type II cancers include high-grade serous, endometrioid, or undifferentiated histotypes, present at late stage (III or IV), grow aggressively, and respond to conventional chemotherapy, but only occasionally to endocrine therapy. Thus, the distinction between Type I and Type II ovarian cancers can inform choice of treatment. 15,16
Whereas Type II serous cancers appear to arise de novo from the walls of ovarian cysts or the surfaces of the ovary or fallopian tube, Type I low-grade serous cancers can grow from noninvasive serous “borderline” tumors of low malignant potential in 60% of cases. High-grade Type II ovarian cancers respond to primary chemotherapy with carboplatin and paclitaxel in approximately 70% of cases. Low-grade serous tumors are resistant, but not refractory, to primary platinum-based therapy. Recurrent low-grade serous ovarian cancer has a very low rate of response. 17 Low-grade mucinous and clear cell histotypes respond to conventional chemotherapy in only 26% and 15% of cases, respectively. 2,17
Low-grade serous cancers exhibit a relatively normal karyotype with wild-type TP53 and BRCA1/2, but exhibit frequent mutations in KRAS genes in 19% to 54% of cases. Low-grade serous cancers express the insulin-like growth factor receptor, and the majority overexpress the IGF-1 growth factor, providing a potential target for therapy. Frequent mutations of KRAS are found in mucinous cancers and in adjacent borderline tumors, consistent with the mutated gene driving malignant progression. A similar pattern of gene expression has been observed in clear cell and low-grade endometrioid carcinomas, consistent with a common cell of origin. 18 Similar mutations have been found in both histotypes. Inactivating mutations of ARID1A, a chromatin remodeling gene, have been reported in 49% of ovarian clear cell carcinomas and 30% of endometrioid ovarian cancers. 19,20 Mutations of PPP2R1A, the regulatory subunit of a serine-threonine phosphatase required for chromosome segregation, have been found in 7% of clear cell ovarian cancers. 19 Phosphatidylinositol-3-kinase (PI3K) signaling is activated in low-grade endometrioid cancers through inactivating mutations and epigenetic silencing of PTEN and activating mutations of PIK3CA. In nonepithelial granulosa cell tumors, recurrent single base mutations (402C-G) of FOXL2, a transcription factor implicated in granulosa cell differentiation, have been found in 97% of cases. 21
Thus, low-grade Type I cancers appear to be driven by mutations that activate Ras/MAP and PI3K signaling in the context of a relatively normal karyotype with wild-type TP53 and BRCA1/2. By contrast, Type II high-grade serous cancers exhibit numerous copy number abnormalities with frequent amplifications and deletions, but with mutations in a very limited number of genes including TP53 and BRCA1/2. The Cancer Genome Atlas Research Network (TCGA) analyzed copy number abnormalities in 489 high-grade serous ovarian cancers, detecting amplification of more than 30 growth stimulatory genes. 22 Amplification and overexpression of genes in the PI3K family occur in more than 50% of Type II cancers, activating the PI3K pathway and conferring “PI3Kness.” 23 In the absence of germline abnormalities of BRCA1/2, homologous DNA repair can be compromised by somatic mutations of BRCA1 and BRCA2 within the cancer alone, BRCA2 can be silenced, and upstream mutations can downregulate BRCA function combined with mutations in other genes potentially involved in homologous recombination, producing “BRCAness” in up to 50% of patients with Type II cancers. 24 DNA sequencing of exons from 316 cancers detected mutations of TP53 in 96%, most of which appeared to be inactivating. 22 Of the 26,000 genes, only a few were mutated in 2% to 4% of cases, including NF1, Rb1, BRCA1, BRCA2, and CDK12. Unlike Type I cancers, fewer than 1% of Type II cancers had mutations of BRAF, PI3KCA, KRAS, or NRAS. Despite the low prevalence of Rb1 mutations, dysfunction of the Rb pathway was found in 67% of high-grade serous cancers. As in the case of epithelial cancers at other sites, both TP53 and Rb were inactivated in two thirds of Type II ovarian cancers. As indicated earlier, low-grade tumors have a high frequency of mutations in PIK3CA, KRAS, ARID1A, and other putative oncogenes and tumor suppressor genes, whereas high-grade ovarian cancers are characterized by mutations in TP53 and BRCA1/2 and marked alterations in DNA copy number. These distinct molecular characteristics indicate that interconversion from Type I to Type II cancers is either a very rare event or does not occur, and thus Type I and Type II tumors likely represent independent diseases. Embracing this concept and performing independent clinical trials and tailored therapy for Type I and Type II tumors will be necessary to improve patient outcomes.
Other Prognostic Subtypes Based on Gene Expression
The pattern of gene expression has been used to identify prognostic subgroups. The Australian Ovarian Cancer Study Group profiled 285 serous and endometrioid tumors to identify six molecular subtypes. 25 Two subtypes were associated with low malignant potential serous tumors and low-grade endometrioid ovarian cancers, whereas the other four transcriptional profiling based subtypes included high-grade serous and endometrioid histotypes including a “mesenchymal” subtype. The immunoreactive subtype expressed T-cell chemokine ligands CXCL11 and CXCL10 and the receptor CXCR3, consistent with a higher level of infiltration by leukocytes. Importantly, lymphocytic tumor infiltration has been associated with an improved outcome. Cases in the proliferative cluster exhibited high expression of the HMGA2 and SOX11 transcription factors, low expression of MUC1 and MUC16 mucins, and high expression of proliferation markers such as MCM2 and PCNA. Differentiated cases have high expression of MUC16, MUC1, and the secretory fallopian tube marker SLPI. Mesenchymal cancers were associated with high expression of HOX genes and markers for stromal components including myofibroblasts (FAP) and microvascular pericytes (ANGPTL2 and ANGPTL1). These subtypes were not associated with changes in overall survival, although a prognostic signature was developed that included 193 genes. Subsequent analyses developed a prognostic “Classification of Ovarian Cancer” (CLOVAR) using gene expression that distinguished groups with markedly different median survival (23 vs. 46 months) and resistance to platinum therapy (63% vs. 23%). 26 Although these classifications can help to stratify future trials, profiles with higher positive and negative predictive value for response to conventional and novel agents will be required in order to affect clinical management.
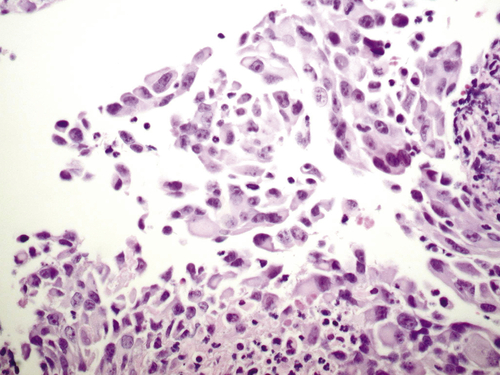
Figure 37-4 Histopathologic features of K-Ras transformed, immortalized human ovarian surface epithelial cells. With permission from Liu, J, Yang G, Thompson-Lanza JA, et al. A genetically defined model for human ovarian cancer. Cancer Res 2004; 64:1655-1663. PMID: 14996724.
Immortalization
Telomerase activity is increased in 80% to 90% of ovarian cancers. Substantially greater telomerase activity is found in ovarian cancers than in borderline lesions or normal ovaries. High-grade serous ovarian cancers demonstrate high degrees of genomic instability and copy number abnormalities, possibly related to bridge-fusion breakage that occurs at telomeric crisis, before the upregulation of telomerase. Immortalization of human ovarian epithelial cells can be achieved by the introduction of human telomerase reverse transcriptase (hTERT) after disruption of the TP53 and/or Rb pathways using SV40T/t antigen or siRNA against Rb. 27 Cells immortalized with SV40 T/t and hTERT can be transformed with mutant Ras, producing cancers resembling low-grade human ovarian cancers that grow as nodules on the peritoneum and exhibit serous papillary histology (Figure 37-4 ). 2 Other combinations of gene aberrations can generate tumorigenic lines from normal ovarian or fallopian tube epithelium, providing a potential approach to explore the roles of specific genomic aberrations in ovarian oncogenesis.
Genomic Abnormalities in Sporadic Ovarian Cancers
A number of genetic and epigenetic abnormalities have been detected in DNA from sporadic ovarian cancers, including amplification, mutation, hypomethylation, chromatin modification, deletion, loss of heterozygosity, and promoter methylation (Table 37-1 ). 2,14,22 High-grade Type II ovarian cancers are genetically unstable both in terms of DNA copy number and in exhibiting a moderate mutation rate (Figure 37-5 ). For unknown reasons, areas of genomic aberration are associated with changes in RNA splicing. The CDK12 gene has been implicated in splicing and is mutated and probably inactivated in 3% of high-grade serous cancers. Aberrant expression of different splicing factors has been observed in ovarian cancer, and altered splicing may affect not only adhesion (CD44), but also the activity of putative oncogenes (EVI-1b and AML-1), as well as the metabolism of xenobiotics (CYP1A1) or of multidrug resistance (SPF45). 2 Processing of miRNAs is also impaired in a fraction of ovarian cancers, with decreased levels of Dicer and Drosha in more than half of ovarian cancers. 28 Low Dicer expression was significantly associated with advanced tumor stage (P = .007) and poor survival (P = .02), whereas low Drosha expression was associated with suboptimal surgical cytoreduction (P = .02). Gene silencing with shRNA, but not siRNA, may be impaired in cells with low Dicer expression. Mutations of Dicer and Drosha are rare in high-grade serous cancer, but mutations of Dicer have been detected in 29% of nonepithelial ovarian cancers. 29 Abnormalities in miRNAs have been found in many different types of cancers related to gain or loss of DNA copies, methylation, or mutation. 30 In ovarian cancers, copy number abnormalities have been found in 37% of 283 loci known to contain miRNAs. 2 A number of miRNAs may act as tumor suppressors and are downregulated in high-grade serous ovarian cancers, including Let-7a/b/d/f and miRs -15/16, -22, -31, -34a/b/c, -125b, -127-3p, -140, -145, -152, -155, -181a, -199a, and -382. 22,30 Conversely, several miRNAs are upregulated and can promote ovarian oncogenesis or chemoresistance, including miRs -15a/16, -20A, -23a/b, -30a/b/c, -92, -93 and -106a, -135b, -141, -200a/b/c, -244, -299-5p, -302d, and -373. Upregulated miRNAs are being evaluated as biomarkers.
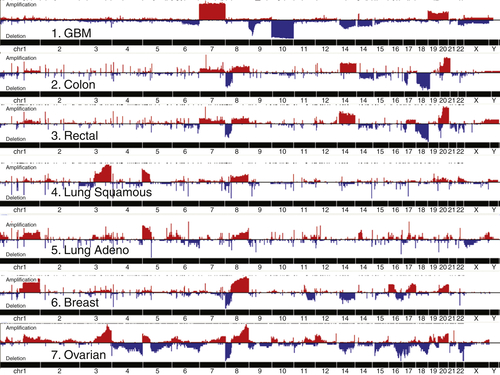
Figure 37-5 Copy number abnormalities in cancers from different sites. GBM, Glioblastoma multiforme. With permission from Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609-615; and Douglas Levine.
Copy number abnormalities (CNAs) are particularly common in epithelial ovarian cancers. Candidate oncogenes and tumor suppressor genes have been mapped to most but not all of the abnormal sites. Amplification of 1q22 includes the RAB25 oncogene. Among the genes encoded on the 3q26 amplicon, protein kinase C iota (PKCiota), SnoN, MDS1-ecotropic viral integration site-1 (mecom/EVI1), and the p110-α catalytic subunit of phosphoinositide-3-kinase (PIK3CA) are overexpressed in a fraction of ovarian cancers and are associated with a poor prognosis. 2 PKCiota protein is required for the establishment and maintenance of epithelial cell polarity. Levels of aberrant PKCiota are markedly increased and/or mislocalized in the majority of serous ovarian cancers and are associated with increased cyclin E protein expression and proliferation. 2 Cyclin E1 amplification and protein levels correlate with a worsened outcome in ovarian cancer. The FGF-1 peptide growth factor encoded by a gene in the amplicon at 5q31 can stimulate cancer growth, stromal growth, and angiogenesis. An amplicon at 8q24 contains c-myc, which is amplified in up to 40% of ovarian cancers, inducing factors required for proliferation and activating telomerase. Another amplicon on chromosome 19q contains the p85 β subunit of PI3K as well as AKT2, a target of PI3 kinase. Another major amplicon at 20q13.2 contains the BTAK/Aurora kinase gene that upregulates c-Myc and activates telomerase.
Loss of tumor suppressor function has been observed in ovarian cancers (Table 37-2 ). 2,14 In some cases, inactivating mutations have been associated with loss of heterozygosity (LOH) (BRCA1, BRCA2, TP53, PTEN), but in others epigenetic changes alone (RASSF1A, DLEC1) or in combination with LOH (ARHI, BRCA1, LOT-1, PEG3, WWOX) have silenced suppressor function. As described earlier, somatic mutation of TP53 is observed in nearly all high-grade type II ovarian cancers. Because of the dominant negative activity of some mutant TP53 protein, TP53 function can be lost with a single genetic event. The pattern of transitions, transversions, and deletions within mutated TP53 genes resembles the pattern of mutations in factor IX deficiency (hemophilia B) in the germline that is thought to be related to spontaneous deamination during DNA replication. 5 If spontaneous mutation during proliferation is a critical mechanism driving carcinogenesis, genetic events requiring only a “single hit” may be favored.
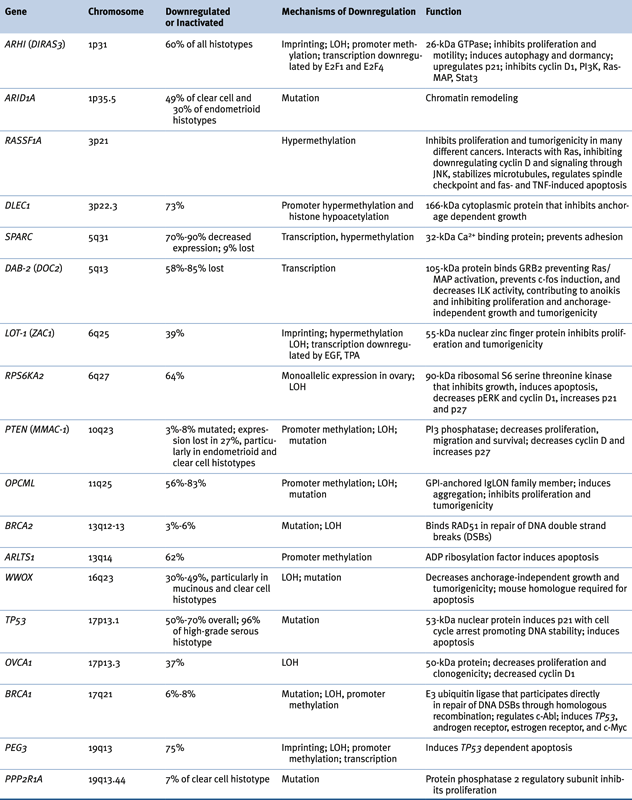
Table 37-3
Oncogenes Associated with Epithelial Ovarian Cancer 14,22
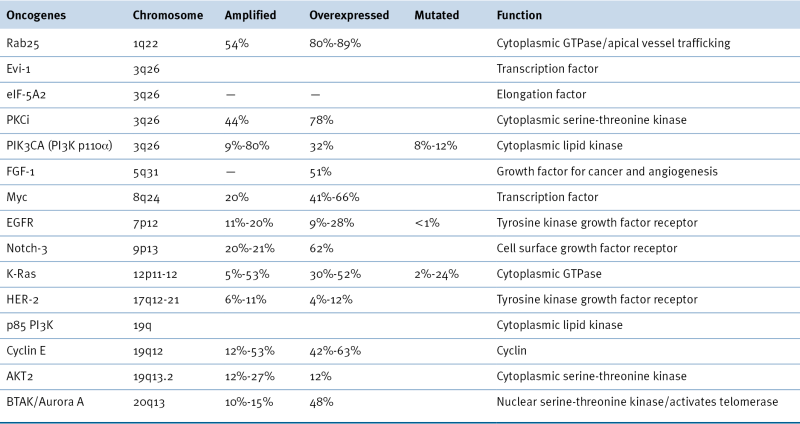
Additional targetable genes with low or high gain of copy number in >20% of high-grade serous ovarian cancers include AKT1, AKT3, CDK2, IL8RB, EPCAM, ERBB3, FGFR2, HDAC4, HSP90AB1, HSP90B1, IGF1, IGFR1, LPAR3, MAP3K6, MAPK15, MAPKAPK2, MAPKAPK5, MECOM, MSTN, MTOR, NCAM1, NOS1, NOS3, PIK3CD, POLB, POLE, RHEB, RICTOR, PPS6KC1, RAPTOR, SKI1, STAT1, STAT4, TERT, TGFB1, TGFB2, TGFBR3, TNFRSF9, VEGFA. 22
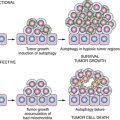
Function of imprinted growth regulatory genes can also be lost in a single genetic or epigenetic event. Approximately 70 human genes are imprinted with only one allele expressed at conception, during embryonic development, and in each normal adult cell. Silencing of the maternal or paternal allele is inherited epigenetically. Among the candidate tumor suppressor genes whose function is downregulated in ovarian cancer (see Table 37-2), at least four are imprinted: ARHI (DIRAS3), LOT-1, NDN, and PEG3. ARHI encodes a 26-kDa GTPase that has 50% to 60% homology to Ras and Rap and that is downregulated in more than 60% of ovarian cancers, associated with decreased time to progression. 31 Expression can be downregulated by multiple mechanisms including LOH, methylation and silencing of the functional allele, transcriptional regulation with E2F1 and E2F4, and shortened mRNA half-life. 32 Re-expression of ARHI at physiologic levels inhibits clonogenic growth and motility of ovarian cancer cells that lack its expression, inhibiting STAT3 translocation, downregulating cyclin D1 and inducing p21WAF1/CIP1 and p27KIP1, producing G1 arrest. ARHI reexpression decreases levels of HIF1-α and inhibits signaling through Ras/MAP and PI3K/TSC2/mammalian target of rapamycin (mTOR), inducing autophagy and tumor dormancy. 33 Among the other imprinted tumor suppressor genes, LOT-1 is a zinc finger protein that presumably acts as a transcription factor 2 and PEG3 is required for TP53-induced apoptosis. 2 Consequently, imprinted genes may regulate the proliferation, motility, and survival of ovarian epithelial cells through multiple mechanisms.
Activation of oncogenes occurs through amplification, overexpression, or mutation in ovarian cancers (Table 37-3 ). 2,14 The two most common aberrations, mutations of members of the PI3K/AKT pathway and of the Ras/RAF pathway, are discussed in the following sections. The FOXM1 and NOTCH pathways are also aberrant in high-grade serous ovarian cancers, albeit at a lower frequency. Abnormalities of receptor and nonreceptor kinases have also been documented.
In contrast to breast cancer, where HER-2 is amplified and overexpressed in 20% to 30% of cases, HER-2 overexpression was found in only 11% of 837 epithelial ovarian cancers in a Gynecologic Oncology Group trial in which trastuzumab produced objective responses in only 7% of tumors with HER-2 overexpression. 2 Unlike lung cancer, the tyrosine kinase domain of EGFR is rarely mutated in ovarian cancer, but the receptor can be amplified in up to 20% of cases. A characteristic constitutively activating deletion in the extracellular domain of EGFR (EGFRvIII), first demonstrated in glioblastomas, has also been detected in a small fraction of ovarian cancers. 2 Constitutive activation of EGFR has, however, not been detected in ovarian cancer cell lines. Inhibition of the EGFR with erlotinib and gefitinib produced only modest clinical activity in ovarian cancer, where objective regression has been observed in 4% to 6% of cases.
Cyclin E maps to an amplicon that is found in a significant minority of ovarian cancers. Knockdown of neighboring genes has shown that CCNE1 is a driver required for clonogenicity that is associated with chemoresistance. 2 Proteomic studies using reverse phase protein analysis have shown that overexpression of CCNE1 protein marks a distinct cluster of cases with poor prognosis.
BTAK/Aurora A kinase, a serine-threonine kinase required for chromosome segregation and centrosome function, is amplified in 10% to 25% and activated in 48% of ovarian cancers. 2 BTAK/Aurora A kinase regulates telomerase activity. Forced expression of Aurora A induces centrosome amplification, cell cycle progression, and chemoresistance to cisplatin and paclitaxel mediated through AKT in a TP53-dependent manner. Chemical inhibition of BTAK/Aurora A kinase downregulates NF-κB, Bcl-XL, and Bcl-2 and enhances sensitivity to chemotherapy. Another cytoplasmic serine-threonine kinase, salt-induced kinase 2 (SIK2), localizes to centrosomes and phosphorylates c-NAP1, permitting centrosome splitting. 34 SIK2 is overexpressed in 30% of ovarian cancers associated with a worse prognosis. Downregulation of SIK2 enhances sensitivity to paclitaxel.
RAB25, a member of the RAB family of small G-proteins implicated in apical vesicle trafficking and polarity, is amplified and overexpressed in approximately half of ovarian cancers and is associated with a poor prognosis. Forced expression of RAB25 protein markedly increased anchorage-dependent and anchorage-independent proliferation; prevented apoptosis and anoikis, including that induced by chemotherapy; and enhanced growth in xenografts. 2 Inhibition of apoptosis was associated with a decrease in the expression of the pro-apoptotic molecules BAK and BAX, as well as activation of the PI3K/AKT pathway. RAB25 is sufficient to increase cellular metabolism and render cells resistant to acute metabolic stress, demonstrating a link between polarity, vesicle trafficking, and cellular metabolism.
In addition to the kinases, growth factors, and signal transducing proteins, members of the family of eukaryotic initiation factors (eIFs) have been implicated in ovarian oncogenesis. The initiation factor eIF-5A2 maps to a region on 3q26 that is amplified in ovarian cancers. Overexpression of eIF-5A2 is associated with advanced-stage ovarian cancer. 2 Forced expression of eIF-5A2 stimulates and antisense inhibits ovarian cancer growth and tumorigenicity.
Several transcription factors are also overexpressed in ovarian cancers. The c-Myc gene is amplified in up to 40% of ovarian cancers, and protein levels are increased in approximately one third of cases, including clear cell and endometrioid histotypes. 2 The c-Myc protein induces E2F1, E2F2, E2F3, and telomerase while blocking TP53-mediated transcription of p21. Despite the probable importance of c-Myc amplification in oncogenesis, no apparent association with survival has been found. A number of approaches to target cells with c-Myc aberrations are beginning to emerge. The association of cMyc with metabolism and particularly in the role of glutamine in cancer biology provides a potential avenue to therapy. The Hippo pathway translational co-activator YAP has also been implicated as a possible oncogene in ovarian cancer that can drive progression and induce resistance to chemotherapy. 35
Aberrant Signaling
Several tyrosine kinase growth factor receptors can be activated in epithelial ovarian cancers, including EGFR, HER-2/ HER-3, M-CSFR, IGFRI, FGFR4, and PDGFR. Binding of relevant ligands to one or more of these receptors can activate signaling through Ras/MAP, PI3K, JNK, JAK/STAT, PKC, and PLCγ or PLC-x with a concomitant alteration in calcium fluxes. Among these several signaling pathways, the Ras/MAP and PI3K pathways appear to be particularly important in regulating the growth, survival, metastatic potential, and drug resistance of ovarian cancer cells. Although the RAS/MAPK and PI3K pathways exhibit few mutations in high-grade serous ovarian cancers, the pathways are frequently activated. The PI3K pathway may be activated by changes in copy number of multiple pathway components as well as by loss of PTEN and INPP4B. Indeed, in the TCGA dataset, the RB1 pathway was dysregulated in 67% of high-grade serous cancers and the PI3K pathway in 45%. 22 The NOTCH signaling pathway was dysregulated in 22%. Stimulation of FGFR4 by FGF-1 activates mitogen-activated protein kinase (MAPK), nuclear factor-κB (NF-κB), and the WNT signaling pathways. 36
In many tumor types, K- or H-Ras is activated by mutations at codons 12 or 61, permitting constitutive binding of GTP rather than GDP to the Ras protein. Activated Ras can signal through the MAPK, PI3K, or Ral pathways, affecting proliferation, survival, motility, invasion, and drug resistance. As noted earlier, the Ras gene is mutated in approximately half of Type I low-grade serous cancers, but rarely in Type II high-grade cancers. Nevertheless, Ras activity as assessed by GTP binding is increased in many Type II high-grade cancers.
The PI3K pathway regulates survival, proliferation, motility, angiogenesis, glucose metabolism, and drug resistance. Activation of the PI3K pathway is observed in 50% of ovarian cancers through multiple mechanisms including amplification or activating mutation of the PI3K p110 α, activating mutation of the PI3K regulatory subunit p85, inactivating mutation of the PTEN phosphatase and amplification of AKT2 (see Table 37-3). 2 The p110 subunit of PI3K is amplified in 9% to 22% of ovarian cancers, overexpressed in 32%, and mutated in 9% to 12%, predominantly in low grade. 2 The pattern of PIK3CA mutation is histotype dependent, being common in endometrioid and clear cell tumors, but rare in serous cancers. The AKT serine-threonine kinase is generally activated physiologically by the products of PI3K. However, in 12% to 27% of ovarian cancers, the AKT2 kinase gene is amplified and is associated with an increase in AKT kinase activity. 2 The p70S6 kinase is downstream of both PI3K and AKT kinase in the signaling cascade. The p70S6 kinase may have a critical role in modulating drug resistance in that rapamycin, an inhibitor of p70S6 kinase, can potentiate sensitivity of some ovarian cancer cell lines to cisplatin-induced apoptosis. 2 Inhibition of activated PI3K decreases cell growth, induces apoptosis, and potentiates paclitaxel chemotherapy. The PI3K pathway is being explored as a therapeutic target in many cancer lineages, including ovarian cancer.
Normal ovarian surface epithelium expresses small amounts of macrophage colony stimulating factor (M-CSF, CSF-1), but little if any of fms, the tyrosine kinase receptor for this ligand. At least 70% of ovarian cancers express and secrete substantially greater amounts of M-CSF, and approximately 50% of cancers express the fms receptor. 2 Interaction of M-CSF with fms stimulates invasiveness and upregulates urokinase-like plasminogen activator (uPA). Expression of uPA correlates with tumorigenicity of ovarian cancer cell lines in xenograft models. 2
Among the nonreceptor tyrosine kinases, the Src tyrosine kinase can be physiologically activated in ovarian cancer cell lines, but the Src gene is rarely amplified or mutated in surgical specimens. 2 Stat3 is phosphorylated by the Janus kinase (JAK) and Src kinase. Phosphorylated Stat3 forms dimers that can be translocated to the nucleus, binding DNA and inducing transcription of genes required for proliferation (cyclin D, c-Myc, c-Fos), survival (Bcl-2, Bcl-XL, XIAP, survivin), and angiogenesis (VEGF). Stat3 can be activated on the intracellular domains of peptide growth factor receptors (EGFR) and cytokine receptors (IL-6R) or by direct interaction with either Src or Abl. Activated pStat3 has been detected in 86% of 322 ovarian cancers. 2 Nuclear localization of pStat3 has been observed in 71% of cases, associated with decreased overall survival. Autocrine and paracrine stimulation with IL-6 activates Stat3, increasing both proliferation and motility. Inhibition of JAK inhibits IL-6–stimulated chemotaxis toward serum and haptotaxis toward fibronectin. 2 Knockdown of Stat3 with siRNA inhibits motility and prevents translocation of Stat to focal adhesions. The imprinted tumor suppressor gene ARHI inhibits proliferation and motility, binds to Stat3, and sequesters it in the cytoplasm, preventing translocation to the nucleus and to focal adhesions. 2 In addition, ARHI can prevent binding of Stat3 to DNA Stat Response Elements in promoter regions.
The endothelin A peptides (ET-1, ET-2, and ET-3) are potent mitogens for several human tumors. ET-1 and its ETA receptor (ETAR) are overexpressed in primary and metastatic ovarian cancers, providing the potential for autocrine stimulation. 2 Interaction with ET-1 also transactivates EGFR, stimulates proliferation, blocks apoptosis, activates integrin-like kinase (ILK), upregulates matrix metalloproteinases (MMPs), and increases vascular endothelial growth factor (VEGF) expression, enhancing angiogenesis.
Lysophosphatidic acid (LPA) is produced constitutively by mesothelial cells and some ovarian cancer cells and accumulates at high levels in the ascites of nearly all ovarian cancer patients. 37 LPA can be detected in the plasma of most patients at all stages of disease. The LPA-2 and LPA-3 receptors are markedly upregulated in ovarian cancers. 2 Interacting with these receptors, LPA stimulates calcium influx, proliferation, motility, chemotaxis, invasion, and resistance to chemotherapeutic agents, signaling through the RAS/MAP and PI3K pathways. LPA is a potent inducer of VEGF, IL8, IL6, and gro, implicating LPA in the accumulation of ascites, neovascularization, and metastasis. LPA can cross activate the EGFR receptor family and other receptor tyrosine kinases, potentially contributing to the activity of these receptors in ovarian cancer. Both the enzyme producing LPA, autotaxin, and the enzymes degrading LPA (LPPs) are aberrant in ovarian cancer. Drugs targeting LPA production and action are potent inhibitors of metastasis.
All three TGF-β isoforms are expressed by normal ovarian surface epithelial cells and regularly inhibit their proliferation, maintaining autocrine growth inhibition. 2 Loss of expression of TGF-β or loss of responsiveness to the growth inhibitory factor is detectable in a fraction of ovarian cancers. Moreover, TGF-β can stimulate the motility and invasiveness of transformed cells. Although mutation in Smad4 is observed in a fraction of ovarian cancers, both TGF-βRI and TGF-βRII receptors are generally intact, as is Smad signaling. 2 Loss of growth inhibition and increased invasiveness may relate to EVI-1 overexpression that is observed in 43% of ovarian cancers 2 and is thought to inhibit transcription of TGF-β–responsive genes. SnoN and AML1, which bind to and regulate Smads, are also aberrant in ovarian cancer.
Müllerian inhibition substance (MIS) bears homology to TGF-β, binds to a receptor with similar structure and function, and is produced by the Sertoli cells of the testis and granulosa cells of the ovary. 2 During embryonic development of gonadal structures, MIS induces atrophy of the Müllerian duct in male mammals. MIS inhibits growth of human epithelial ovarian cancer cells in culture and in xenografts. Binding of MIS to MISII G-protein–coupled receptors upregulates p16, produces G1 cell cycle arrest through an Rb-independent mechanism, and induces apoptosis. Some 56% of human ovarian cancers express MISII receptors, and clonogenic growth can be inhibited with MIS in more than 80% of cancers bearing receptors. MIS enhanced the anticancer activity of suboptimal doses of chemotherapeutic agents against human and murine ovarian cancer cell lines in culture and as xenografts. 2
Stem Cells
Both normal and malignant tissues are thought to contain small subpopulations of stem cells with unlimited replicative potential that can be passed serially from mouse to mouse in vivo or as spheroids in cell culture. In cancers, tumor-initiating cells that have stem cell–like characteristics are generally resistant to chemotherapy and radiotherapy. The phenotype of human ovarian cancer stem cells remains to be fully defined. Increased tumor-initiating potential has been associated with CD133+ALDH1+ and CD44+CD117+ subpopulations in ovarian cancer cell lines and clinical specimens. 38 CD133 has been associated with stem cells from normal and malignant tissues arising at multiple sites. Aldehyde dehydrogenase mediates resistance to certain drugs and toxins. CD44 is the hyaluronate receptor required for adhesion, and CD117 is the transmembrane tyrosine kinase growth factor receptor, c-Kit. CD24 can be associated with both subpopulations and marks ovarian cancer cells with tumor-initiating capacity and chemoresistance. CD24+ ovarian cancer cells express a number of stem cell biomarkers including Nestin, Beta-catenin, Bmi-1, Oct3/4, Notch1, and Notch4. Recent studies suggest that normal ovarian stem cells occupy a niche in the hilus of the mouse ovary at the junction between the ovarian surface epithelium, the peritoneal mesothelium, and the tubal (oviductal) epithelium. The cells cycle slowly and express a number of stem cell markers including ALDH1, LGR5, LEF1, CD133, and CK6B. Spheroids can be passaged serially in culture, and cells can be transformed by inactivating TP53 and Rb1. 39
Animal Models
Among the animal models, spontaneous ovarian cancers occur in approximately 40% of egg-laying hens 4 years of age. 40 Some 46% of ovarian cancers have mutations of TP53, and the majority express MUC16. Chickens have been used to test different hormonal strategies for preventing epithelial ovarian cancer.
Murine ovarian epithelial cells have been engineered to express different combinations of oncogenes and tumor suppressors. 40 When target cells were derived from transgenic mice that lacked TP53 expression, the addition of any two of three oncogenes—c-Myc, activated K-Ras, and activated AKT—were sufficient to induce high-grade cancer in ovarian surface epithelial cells. In mice that were deficient for both TP53 and BRCA1, Myc overexpression is sufficient to induce cancers. In a serous murine ovarian cancer model, disruption of either Rb or TP53 produced relatively few ovarian cancers, whereas disruption of both Rb and TP53 produced a greater number of high-grade epithelial ovarian cancers, albeit with long mean latency (227 days). High-grade ovarian epithelial cancers were induced in a third model when SV40 T antigen, which blocks both TP53 and Rb, was driven by the Müllerian hormone type 2 receptor (Ambr2) promoter. A model for low-grade ovarian cancer has been generated in mice in which the PTEN gene was disrupted and an oncogenic form of KRasG12D was expressed selectively in ovarian surface epithelial cells, using mice in which Cre recombinase was driven by the Amhr2 promoter. Finally, high-grade serous cancers of the fallopian tube have developed in in mice where PTEN and Dicer were conditionally depleted using the Amhr2-Cre mouse strain. Consequently, ovarian cancers have been associated with loss of TP53, Rb, PTEN, and Dicer function, as well as activation of Ras, AKT, and Myc. To date, however, murine models have not succeeded in mimicking the abnormal DNA copy numbers observed in human ovarian cancers, possibly related to the telomere length in murine cells and relative resistance to telomeric crisis.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


