PMS2, MSH6
BMPR1A
PTEN
Hereditary Colorectal Cancer Syndromes and Molecular Pathways of Colorectal Carcinogenesis
It is now generally accepted that the accumulation of somatic mutations and epigenetic defects drives the initiation and progression of tumor development. The constellation of somatic mutations that commonly accumulate in a clonal fashion in CRCs has largely been defined. 17–20 However, only a limited subset of this collection of gene defects is usually present in any individual tumor. Thus far, the somatic mutations of greatest initial interest to the field have been those that are recurrent and clonal (i.e., present in all, or nearly all, neoplastic cells of a primary tumor at a given stage, but not present in the normal cells of the patient). It is inferred that such recurrent, clonal mutations are causal in promoting further tumor outgrowth/progression, because somatic mutations can only become clonal by a limited number of mechanisms. The genetic alteration itself could have been selected for because it provided the cell with a growth advantage, allowing it to become the predominant cell type in the tumor (clonal expansion). Alternatively, the specific alteration detected might have arisen coincident with another, perhaps undetected, alteration that was the crucial change underlying clonal outgrowth.
As noted previously, only about 5% of all CRC cases are associated with defined highly penetrant cancer syndromes. The bulk of these cases are attributable to hereditary nonpolyposis colorectal cancer syndromes (HNPCCs), with another significant subset associated with the familial adenomatous polyposis (FAP) syndrome (Table 34-1 ). A few other syndromes comprise the remaining highly penetrant CRC syndrome cases.
Adenomatous polyposis coli Gene: Gatekeeper in Familial Adenomatous Polyposis and Sporadic Cancer
Familial Adenomatous Polyposis
FAP is an autosomal dominant syndrome affecting about 1 in 8000 individuals in the United States and accounting for about 0.5% of CRCs. 4,5 Hundreds to thousands of adenomas arise in the large bowel and rectum beginning in the second decade of life. Although only a fraction of the adenomas may progress to cancer, the lifetime incidence of colorectal cancer in untreated FAP patients approaches 100% with a mean age at diagnosis of 39 years, necessitating the prophylactic removal of the patient’s colon early in adult life. 4 The gene that when mutated underlies FAP is the adenomatous polyposis coli (APC) tumor suppressor gene on chromosome 5q21. Germline mutations have been identified in one APC allele of affected individuals in 90% to 95% of families with FAP studied. More than 95% of mutations lead to premature truncation of APC protein synthesis—about two thirds by small insertions or deletions leading to a frameshift, and the remainder by introducing a stop codon 21 (Figure 34-1 ). Up to 25% of FAP cases seem to be caused by de novo germline mutations and therefore do not show the characteristic autosomal pattern of inheritance. 4 Moreover, about 20% of individuals with a de novo APC mutation display somatic mosaicism for the mutation in their tissues, with the mutant APC allele present in only subsets of cells within a given tissue and/or in germ cells. 4 Some of these cases with somatic mosaicism may manifest milder polyposis phenotypes than individuals carrying inherited germline APC mutations, as the defective APC allele is present in only a fraction of cells in the somatic mosaicism situation.
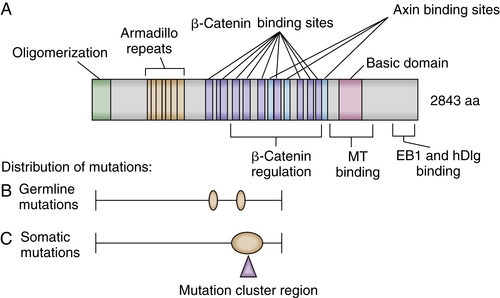
Figure 34-1 Schematic representation of the adenomatous polyposis coli (APC) protein and mutation histograms (A) Selected sequence motifs of the 2943-amino-acid APC protein and its interaction partners are indicated. The amino-terminal region has a domain that regulates its oligomerization. Repeated sequences with similarity to the Drosophila armadillo protein and its vertebrate homologue β-catenin (so-called armadillo repeats) are localized in the amino-terminal third of APC. Multiple independent 20-amino-acid repeats mediating binding to β-catenin and several binding sites for the Axin protein (termed SAMP repeats) are localized in the central third of APC. The carboxy-terminal third contains a basic region that is involved in microtubule (MT) binding and mediates interactions with the proteins EB1 and hDlg. (B) The frequency and distribution of germline mutations in familial adenomatous polyposis (FAP) patients are indicated with respect to the APC coding region. Virtually all mutations result in premature truncation at or very close to the mutation position. Two apparent mutational hotspots are seen at codons 1061 and 1309. (C) Frequency and distribution of somatic APC mutations in sporadic colorectal cancers are indicated. The mutations appear to predominate in the “mutation cluster region” (MCR), and mutations at codons 1309 and 1450 are most common. (A-C with permission from Polakis P. Mutations in the APC gene and their implications for protein structure and function. Curr Opin Genet Develop 1995;5:66).
Several extracolonic tumors and symptoms are associated with FAP (see Table 34-1). The combination of polyposis with brain tumors (in particular, medulloblastoma in pediatric cases) has been termed Turcot syndrome. 22 Gardner syndrome comprises extensive polyposis with epidermoid cysts, desmoid tumors, and osteomas and seems to be correlated with mutations between APC codons 1403 and 1578. 23,24 Upper gastrointestinal polyps are responsible for a large proportion of morbidity and mortality in FAP patients after prophylactic total colectomy. This is particularly true for the duodenal cancers that develop in 4% to 12% of patients. 4,25,26 Benign gastric fundic gland polyps and gastric adenomas, potential premalignant precursor lesions of gastric cancer, are observed with increased frequency, ultimately leading to stomach cancer in 0.5% of cases. 4,25,26 In only a small percentage of FAP cases, thyroid cancer, bile duct cancer, hepatoblastoma (pediatric), and central nervous system tumors such as medulloblastoma are observed. In addition to FAP, rare germline APC variants appear to play a role in other familial CRC cases. 27 Epidemiologic studies have revealed that the APC I1307K (isoleucine-to-lysine substitution at codon 1307) allele is present only in individuals of Ashkenazi Jewish origin, and those who carried the I1307K allele had a roughly twofold increased risk of developing CRC. 27 The mechanism underlying increased risk of adenomas and CRC in carriers of the APC I1307K allele is that the allele appears to be more susceptible than the wild-type APC allele to somatic mutations—particularly insertion and deletion of one or a few nucleotides near the homopolymeric adenine tract created by the lysine 1307 codon substitution, resulting in frameshift mutations. 27
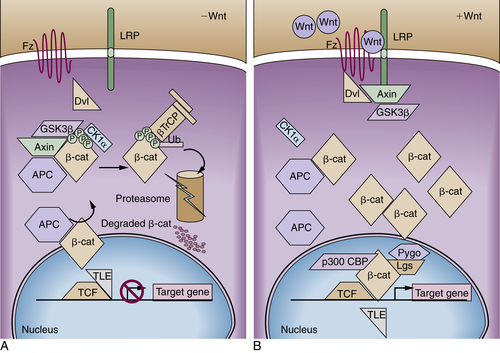
Figure 34-2 Model of adenomatous polyposis coli (APC) and β-catenin function (A) The APC protein is part of a “destruction complex” containing glycogen synthase kinase 3β (GSK3β), Axin, and casein kinase 1α (CK1α), which phosphorylates β-catenin in conserved serine and threonine residues in the N terminus. This phosphorylation creates an epitope for recognition by the F-box protein β-TrCP as part of a ubiquitin ligase complex, which leads to polyubiquitination and proteasomal degradation of β-catenin. Hence, in most cells free β-catenin levels are kept low and transcription of target genes is repressed by recruitment of repressors of the transducin-like enhancer of split family (TLE). (B) On binding of Wnt ligands to a cognate receptor complex consisting of one member of the Frizzled (Fz) family and one member of the low-density-lipoprotein receptor-related protein family (i.e., LRP5 or LRP6), the destruction complex is disassembled in part by recruitment of Axin to the LRP receptors and in part by not clearly defined action of disheveled proteins (Dvls). Similarly inactivating mutations of the APC protein or the Axin1 protein or mutations of the conserved phosphorylation sites of β-catenin lead to the accumulation of free β-catenin, which enters the nucleus and activates transcription of target genes by displacement of co-repressors and recruitment of coactivators such as CBP/p300 and a complex of the homologues of Drosophila Legless (Lgs) and Pygopus (Pygo).
Somatic APC Mutations in Sporadic Tumors
Notwithstanding the critical role of the APC gene in FAP and related variant syndromes, the APC gene has an even more prominent role in sporadic colorectal tumors, as about 80% of sporadic colorectal adenomas and carcinomas have somatic mutations inactivating APC, 28 the overwhelming majority of which lead to premature truncation of the APC protein. APC mutations have been found in a number of the earliest sporadic lesions analyzed, including microscopic adenomas composed of only a few dysplastic glands. 29 As predicted by Knudson’s two-hit model for tumor suppressor genes, both APC alleles appear to be inactivated in the majority of colorectal adenomas and carcinoma, as a result of localized mutations in both alleles or localized mutation in one allele and chromosomal loss of the remaining allele. 28
APC Protein Function
The APC tumor suppressor gene encodes a large protein of approximately 300 kDa (see Figure 34-1). The APC protein likely has multiple functions in the cell, 30 but the best understood function of APC is as a major binding partner and regulator of the β-catenin protein. 31 β-Catenin was first identified because of its role in linking the cytoplasmic domain of the E-cadherin cell-cell adhesion molecule to the cortical actin cytoskeleton, via binding to the adaptor molecule α-catenin. Based on studies from many different investigators, a model has been developed to explain the biologic significance of APC’s interaction with β-catenin. 31 The model proposes that in the absence of an activating Wnt ligand signal, APC binds to and collaborates with the scaffold protein Axin to promote sequential phosphorylation by casein kinase I and glycogen synthase kinase-3β (GSK3β) of several conserved serine/threonine residues in the N-terminal region of β-catenin, thereby targeting β-catenin for ubiquitination and proteasomal degradation (Figure 34-2 ). In a physiologic setting, the activating Wnt ligands inhibit degradation of β-catenin via binding to their cognate receptor complex of a Frizzled and an LRP5/6 protein.
In the roughly 80% of CRCs where both APC alleles are defective, the coordinated destruction of β-catenin is severely impaired, essentially mimicking constitutive activation of Wnt ligand-mediated signaling (see Figure 34-2). As a result, β-catenin accumulates in the cytoplasm, complexes with DNA binding proteins of the TCF (T-cell factor family)/Lef (lymphoid enhancer family) family, and translocates to the nucleus. Once there, β-catenin functions as a transcriptional co-activator, activating the expression of TCF-regulated genes. In a subset of the cancers that lack mutations in APC, somatic mutations in β-catenin have been found. 32 A critical net consequence of β-catenin stabilization by APC inactivation or other somatic mutations in CRCs is that the constitutive activation of β-catenin/TCF transcription appears to promote a stem or progenitor cell phenotype in the affected epithelial cells independent of their position in the crypt compartment. 31 Normally, β-catenin/TCF activation is restricted to the crypt base, especially in the so-called crypt base columnar stem cells that are characterized by expression of the Wnt-regulated Lgr5 gene and protein. 31
Further work has implicated β-catenin stabilization not only in the establishment of a crypt progenitor program but also in the spatial organization and migratory pattern of the cells in the continuous renewal of crypt. 31 Strikingly, feedback inhibitors functioning in the Wnt/β-catenin/TCF pathway are prominent among the critical proteins encoded by β-catenin/TCF-regulated genes, such as the AXIN2, DKK1, NKD1, APCDD1, and WIF-1 proteins, all of which function in some fashion to antagonize Wnt signaling. 31 In CRCs with APC mutations, the ability of the induced feedback regulator proteins to interfere with stabilized β-catenin is largely or entirely abrogated, because the induced Wnt-pathway feedback regulators function upstream of or at the level of the APC protein in the pathway. 31 Interestingly, some of the induced Wnt pathway feedback regulators that are highly expressed in CRCs with APC mutations, such as AXIN2, may play important contributing roles in CRC progression, such as through the ability of AXIN2 to interact with other proteins and pathways in the promotion of epithelial-mesenchymal transition and invasive phenotypes. 33
Mouse Models of FAP and Genetic and Epigenetic Modifiers
An important mouse genetic model of intestinal tumorigenesis known as the Min (for multiple intestinal neoplasia) mouse was described more than two decades ago. 34 The Min mouse carries a germline mutation in the murine homologue of the APC gene, resulting in truncation of the murine Apc protein at codon 850. The intestinal tumor phenotype of Min or Apc(Min) mice is similar, but not identical, to that seen in FAP patients, with Apc(Min) mice developing largely small intestinal tumors, in contrast to the colorectal lesions seen in FAP patients.
Other cellular genes can significantly influence the number of polyps that arise in mice that are heterozygous for the Apc(Min) mutant allele or other mutant Apc alleles. For instance, when the Apc(Min) mutation was introduced into mice of varying genetic backgrounds, significant variability in the number of intestinal tumors was seen. This finding was attributable to the effects of strain-specific modifier genes unlinked to the Apc locus, particularly the modifier gene Pla2g2a, which encodes a secreted phospholipase A2 protein. 35 Other genes that substantially modify intestinal tumorigenesis in Apc-mutant mice include genes encoding DNA methyltransferases. Specifically, mice that carry the Apc(Min) mutation and a germline defect in one allele of the maintenance DNA methyltransferase Dnmt1 or a gut-specific deletion of both Dnmt3 alleles have a two- to threefold reduction in macroscopic polyp number. 36,37 Treatment of Apc(Min) mice with 5-azacytidine, a pharmacologic inhibitor of DNA methyltransferases, resulted in a more than fivefold reduction in polyp number. 36 Combining the genetic and pharmacologic manipulations of DNA methyltransferase activity synergistically reduced the polyp number roughly 50-fold. 36 Treatment of mice with NSAIDs that inhibit COX-1 and COX-2 or agents that specifically inhibit COX-2 activity also markedly inhibited adenoma formation. 38,39 The findings highlight the utility of the mouse genetic models for identifying novel genes and pharmacologic agents that modify intestinal tumor development.
Other Forms of Intestinal Polyposis
Some patients with germline mutations in the APC gene have a milder or attenuated FAP (AFAP) phenotype, due to the nature of the germline APC mutation that they carry and/or other genetic variants that they may carry that modify the severity of their polyposis phenotype. 4,5 However, many of the individuals who have an AFAP phenotype and typically present to clinical attention between 40 and 60 years of age with fewer than a dozen to a hundred polyps do not carry a germline APC mutation. 4,5 Rather, these affected individuals carry homozygous mutations in the MYH gene encoding the base-excision repair pathway protein MutYH. The syndrome is often termed MAP for MYH-associated polyposis syndrome. 4,5 The mutations in the MYH gene lead to constitutional defects in the repair of oxidative DNA damage in cells, with increased GC-to-AT base-pair transversions arising. As a result of the increased mutation frequency, in the colon, somatic mutations in various genes likely arise at increased frequency, with the somatic mutations contributing to adenoma formation.
Some individuals and families who develop 10 to 100 or more adenomas during adult life, and who also develop CRC at an increased rate unless managed clinically, have been found to lack germline mutations in the APC or MYH genes. Recent studies have shown that some of these affected individuals and families carry heterozygous germline mutations in the POLD1 and POLE genes encoding DNA polymerases δ and ε, respectively. 40 The mutations appear to affect the proofreading domains of the polymerases. In the case of POLD1 mutations, affected individuals appear to be predisposed not only to adenomas and CRC but also to endometrial cancer and perhaps brain tumors. 40 The colon tumors arising in POLD1 and POLE mutation carriers were found to have inactivated the remaining wild-type copy of the gene by loss of heterozygosity (LOH). As a result, the tumors lack intact polymerases δ or ε function, depending on the nature of the patient’s germline mutation, and the tumors manifest a markedly greater frequency of base-substitution mutations relative to similar stage colon tumors with intact polymerase δ and ε function, presumably as a direct result of the deficits in polymerase δ or ε function. 40
Other intestinal polyposis syndromes in which patients manifest numerous nonadenomatous lesions have also been described. 4,5 Several of these syndromes have an increased risk of gastrointestinal and/or non-gastrointestinal cancers (see Table 34-1). Starting from childhood, patients with the autosomal dominant juvenile polyposis syndrome (JPS) develop multiple hamartomatous polyps throughout the gastrointestinal tract with some preference for the colon and the stomach. JPS is a genetically heterogeneous disease, with inactivating mutations in the SMAD4 and BMPR1A genes among the underlying genetic bases. Cowden syndrome is an autosomal dominant syndrome in which affected individuals develop macrocephaly and hamartomas in many organ sites, including the breast, thyroid, skin, central nervous system, and gastrointestinal tract. 4,5 The gene responsible for Cowden syndrome is the PTEN tumor suppressor gene on chromosome 10q23. Acting as a phospholipid-phosphatase, the PTEN protein is a major antagonist of the phosphatidylinositol-3-kinase (PI3K) survival pathway. In a small number of nonfamilial cases of juvenile polyposis lacking other features of Cowden syndrome, PTEN germline mutations have been reported. 4,5 Peutz-Jeghers syndrome, a rare autosomal dominant condition affecting fewer than 1 in 25,000, is characterized by gastrointestinal hamartomatous polyps and mucocutaneous melanin deposition. 4,5 The hamartomatous polyps contain essentially normal epithelial cells, but the mucosal components are arranged abnormally. Germline mutations in the LKB1/STK11 tumor suppressor gene on chromosome 19p can be seen in a significant fraction of cases. 4,5 Inactivation of this tumor suppressor gene leads to the hyperactivation of the mammalian target of rapamycin (mTOR) pathway, which is responsible for integrating the nutritional supply with cell proliferation and growth. Finally, recent studies of a rare inherited syndrome known as hereditary mixed polyposis syndrome (HMPS), in which individuals are predisposed to develop a range of lesions, including Peutz-Jeghers polyps, juvenile polyps, serrated lesions, conventional adenomas, and CRC, is due in some families to germline mutations that lead to overexpression of the bone morphogenetic protein (BMP) antagonist GREM1. 41 Thus, although HMPS is connected to JPS, based on the fact that BMPs signal through the transforming growth factor β (TGF-β) pathway, further work is needed to understand how derangements in TGF-β and BMP signaling contribute to predisposition to colorectal tumors.
DNA Mismatch Repair Deficiency and Lynch Syndrome
Hereditary nonpolyposis colorectal cancer (HNPCC) was arguably the first inherited cancer syndrome to be well described in the literature. In 1913, Warthin presented a particularly striking example of a three-generation family with CRC and other cancers, including gastric cancer and tumors of the female reproductive tract. 42 Following Warthin’s lead, Lynch and others described kindreds with autosomal dominant patterns of CRC, not accompanied by extensive polyposis. 43 In such families, CRCs of early onset were seen, often along with cancers in some other organs including gastric, uterine endometrial, ovarian, small bowel, renal, and hepatobiliary cancers. 44 In recognition of Dr. Henry Lynch’s major contributions to the field, HNPCC is often referred to as Lynch syndrome.
Criteria for the diagnosis of Lynch syndrome in families have often included the following: affected families must show Lynch syndrome–typical tumors in three relatives (one being a first-degree relative to the other two) in at least two successive generations, with one of the tumors occurring before age 50 (so-called 3-2-1 rule). The sensitivity of these diagnostic criteria is considerably less than 100% for identifying those affected by Lynch syndrome, and the specificity of the criteria is also an issue. 4 However, the diagnostic criteria proved critical for the initial discovery of the germline mutations underlying Lynch syndrome, and the criteria have also been useful in clinical assessment over the past 15 to 20 years since the genetic bases of Lynch syndrome were uncovered. By way of a brief background, the early genetic work indicated genetic heterogeneity for Lynch syndrome, with mapping of the predisposing gene to chromosome 2p in some families 45 and to chromosome 3p in other families. 46 In yet other families with Lynch syndrome, no evidence for linkage to chromosome 2p or 3p was found.
To explore the relevance of Knudson’s two-hit hypothesis for Lynch syndrome genes, investigators initially sought to demonstrate loss of parental heterozygosity for chromosome 2p in cancers from individuals carrying a defect in that particular predisposition gene. 45 However, not only was there no LOH of chromosome 2p sequences in the cancers, but microsatellite DNA sequences examined in the analyses demonstrated marked length variations in tumor tissues compared with the patient’s normal tissue. Microsatellite changes were seen at many different loci scattered throughout the genome and in all tumors from patients with Lynch syndrome. 45 The phenotype was termed the microsatellite instability” (MSI) or replication error (RER) phenotype. Cancers with evidence of microsatellite instability at more than 40% of a panel of mononucleotide and dinucleotide sequences are termed high-frequency MSI (MSI-H) cancers. Although most CRCs display no instability when a panel of microsatellite tracts are studied—so-called microsatellite stable (MSS) cases—a subset of cancers show low-frequency instability of the microsatellite markers, termed MSI-L.
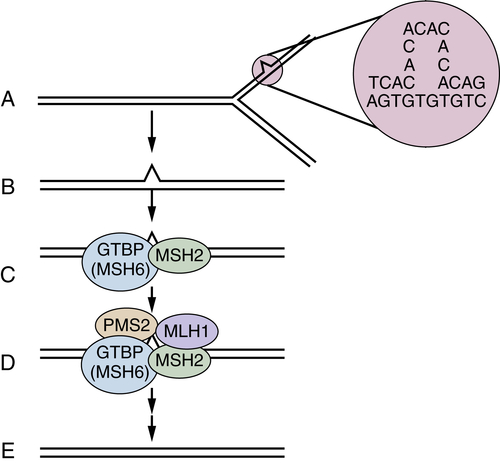
Figure 34-3 Mismatch repair pathway in human cells (A, B) During DNA replication, DNA mismatches may arise, such as from strand slippage (shown) or misincorporation of bases (not shown). (C) The mismatch is recognized by MutS homologues, perhaps most often MSH2 and GTBP/MSH6, although MSH5 may substitute for GTBP/MSH6 in some cases. (D, E) MutL homologues, such as MLH1 and PMS2, are recruited to the complex, and the mismatch is repaired through the action of a number of proteins, including an exonuclease, helicase, DNA polymerase, and ligase. A-E with permission from Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159-170.
The finding of MSI-H in the cancers was noteworthy because similar DNA instability phenotypes had previously been seen in mutant yeast strains with defective DNA mismatch repair genes. The prediction that defects in one or more DNA mismatch repair genes might underlie Lynch syndrome was quickly borne out (Figure 34-3 ). A human homologue of the bacterial mutS mismatch repair gene, designated MSH2, was mapped to chromosome 2p, and one allele was found to be mutated in the germline of a subset of Lynch syndrome patients, with the remaining allele inactivated in cancers arising in the mutation carriers. Other genes involved in DNA mismatch repair were studied and found mutated in other groups of Lynch syndrome patients, including the MLH1 gene on chromosome 3p, the PMS2 gene on 7q, and the GTBP/MSH6 gene on chromosome 2p. 47 Mutations in the MSH2 and MLH1 genes are a far more common cause of Lynch syndrome than mutations in the other mismatch repair genes, with MSH2 and MLH1 mutations together accounting for about 70% to 75% of the known mutations in Lynch syndrome patients. 47
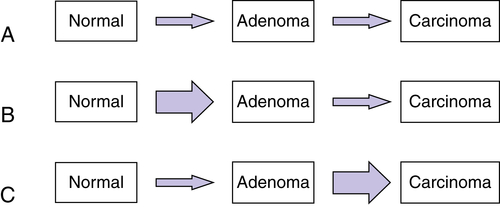
Figure 34-4 Relative effects of germline mutations on tumor initiation and progression (A) In sporadic cancers, both initiation of a neoplastic lesion (e.g., the adenoma) and progression to an advanced lesion (i.e., the carcinoma) are rate-limiting events because two somatic mutations are required for inactivation of tumor suppressors such as adenomatous polyposis coli (APC) (initiation of adenoma) and p53 (tumor progression). (B) In the case of familial adenomatous polyposis (FAP), germline inactivation of one APC allele markedly increases the formation of adenomas, because inactivation of both APC alleles is a critical (rate-limiting) event in adenoma formation, and those with inactivation of one allele in all colonic cells need only inactivate the remaining APC allele to initiate adenoma formation. (C) In the case of Lynch syndrome (hereditary nonpolyposis colorectal cancer [HNPCC]), germline inactivation of one of the mismatch-repair genes (e.g., MSH2 or MLH1) coupled with somatic inactivation of the remaining allele in an initiated lesion (e.g., an early adenoma) greatly increases the mutation rate, and subsequently the rate and speed of progression to more advanced lesions. With permission from Bettingon et al. The serrated pathway to colorectal cancer: current concepts and challenges. Histopathology. 2013; 62:367-386.
In the normal cells of a patient with Lynch syndrome, DNA repair is rarely impaired, because the cells have a wild-type copy of the gene (i.e., the copy inherited from the nonaffected parent). However, during cancer development, the remaining wild-type allele is inactivated by a somatic mutation. Following inactivation of the wild-type allele, full mismatch repair activity is lost (see Figure 34-3). Then, affected cells manifest a mutator phenotype and accumulate mutations in a much more rapid fashion. Lynch syndrome is, therefore, a disease with more rapid tumor progression from a benign, initiated clone to frank malignancy (Figure 34-4 ).
Although germline mutations in the known mismatch repair genes have been detected in only 2% to 4% of CRC patients, roughly 15% of all CRCs display the MSI-H phenotype. 47 The somatic inactivation of MLH1 via epigenetic mechanisms, including hypermethylation of its promoter, seems to be responsible for most MSI-H CRC cases outside of Lynch syndrome families. 47
Many of the mutations that arise in cells with the MSI-H phenotype may be detrimental to cell growth or may exert no positive selection pressure. However, a subset of mutations could potentially activate oncogenes or inactivate tumor suppressor genes. An example of a gene containing a mononucleotide tract in its coding sequence that is frequently mutated in MSI-H CRCs is the TGF-β type II receptor. 47 Some other genes commonly altered in MSI-H CRCs are described later.
Inflammation and Colon Cancer
Chronic inflammatory bowel disease (IBD), especially ulcerative colitis, confers an increased risk of CRC. In the case of UC, the risk of CRC is associated with both the duration and extent of the inflammatory disease. From a pathogenetic standpoint it is notable that UC-associated CRCs often develop without the formation of a polyp as a precursor lesion, although dysplasia is a common factor. 15,48 Reducing the degree of inflammation in IBC seems to reduce the risk for CRC. 49 Some insights into the factors and pathways by which inflammation contributes to colitis-associated CRC (CAC) have begun to emerge 50 and likely include important roles for NF-κB signaling in both immune cells and epithelial cells and local activation of various cytokines and lymphokines, including tumor necrosis factor, interleukin 1 (IL-1), IL-6, IL-17, and IL-23. 50
The responses of the innate and cellular immune systems to variations in intestinal microbial communities as well as inflammatory mediators all seem to have important and potentially much broader contributing roles in intestinal neoplasia beyond the setting of CAC. For instance, a human colonic commensal—enterotoxigenic Bacteroides fragilis—can affect tumor progression in the Apc(Min) mouse model of intestinal tumorigenesis in part via inflammation and the generation of IL-17–expressing T helper 17 [T(H)17] cells. 51 Recent studies have further highlighted the role of microbes and microbial products, along with IL-23 and T(H)17 cells in Apc-mutation–dependent colon tumor progression in the mouse. 52 Future studies are likely to further clarify the likely complex relationships among the intestinal microbiota, host immune cells, and epithelial cells in the initiation and progression of colon neoplasia in mouse models and in humans.
Recurrent Somatic Alterations in Colorectal Cancer
Somatic alterations have key roles in the initiation and progression of colorectal tumors, in patients with inherited predispositions to CRC as well as in the majority of CRC patients whose tumors are apparently sporadic. A sizeable number of mutational alterations in CRCs were identified over the past 30 years, largely by various combinations of genetic and genomic approaches. 18 Among the major targets for somatic mutations in CRCs identified before whole genome sequence-based approaches included oncogenic (activating) point mutations affecting KRAS (about 40% of CRCs), NRAS (about 5%), PIK3CA (20%), and BRAF (5% to 10%), as well as amplifications of the EGFR (5% to 10%), CDK8 (10% to 15%), CMYC (5% to 10%), and ERBB2 (5%) genes. 18 Similarly, among the tumor suppressor genes known to be frequently affected by somatic mutations in CRCs as a result of the “old-school,” focused sequencing approaches were the following: APC (80%), TP53 (60%), FBXW7 (20%), SMAD4 (10% to 15%), PTEN (10%), SMAD2 (5% to 10%), ACVR2A (10%), and TGFBR2 (10%). 18
Over the past few years, comprehensive sequencing approaches have allowed for expansive analyses of the nature and spectrum of mutational alterations in CRCs. 17,19,20 One of the major advances of the comprehensive sequencing-based approaches has been knowledge of the variation in total somatic mutations among CRCs. For example, in an exome-sequence analysis of 224 tumor and normal pairs from CRC patients, it was found that 84% of the tumors had a median number of nonsilent (predicted to change protein-coding sequence) somatic mutations of 58 per tumor; the remaining 16% of the tumors were reported to manifest a “hypermutated” phenotype, with a median of 728 somatic nonsilent mutations per tumor. 19 These findings that about 16% of CRCs had a hypermutated phenotype was not entirely unexpected. As described earlier in the discussion of mismatch repair gene defects in Lynch syndrome and also apparently sporadic CRCs, about 15% of all CRCs manifest the MSI-H phenotype, in the majority of MSI-H CRCs because of epigenetic silencing of the MLH1 gene. 18,47 As a result of their defective mismatch repair pathway function, the MSI-H CRCs were known to have a hypermutable phenotype. However, what was a bit unexpected about the hypermutated CRCs identified via the comprehensive sequencing approaches was that about 25% of the hypermutated tumors lacked the MSI-H phenotype and had intact function of MLH1. Instead, the hypermutated non-MSI-H cases had somatic mutations in other mismatch repair genes (e.g., MSH2, MSH3, MLH3, MSH6, PMS2) or in the POLE gene encoding DNA polymerase ε. 19 One somewhat puzzling issue is why somatic mutations in certain mismatch repair genes do not invariably lead to demonstrable instability in the microsatellite sequences that define the MSI-H phenotype, whereas epigenetic silencing of MLH1 does apparently lead to the MSI-H phenotype in almost all cases. Perhaps part of the explanation might be the relative timing of the defects in mismatch repair function in the natural history of a given CRC.
Besides clarifying the mutation rate in CRCs and illuminating the molecular basis for the markedly increased mutation rate in the roughly 16% of CRCs that are hypermutated, comprehensive molecular characterization efforts have yielded additional insights, such as refinements in the estimated frequencies for known oncogene and tumor suppressor gene defects in CRC, the identification of some new oncogene and tumor suppressor gene defects contributing to CRC, and some initial insights into how epigenetic changes and gene expression patterns may be associated with certain mutation patterns as well as biologic and clinicopathologic subsets of CRC. Table 34-2 summarizes some of the genes most frequently affected by somatic mutations in nonhypermutated and hypermutated CRCs. 19,20
In addition to the extensive data on localized mutation frequencies, comprehensive molecular characterization efforts have begun to further inform understanding of recurrent gene amplification, deletion, and translocation in CRCs. Besides largely confirming the gene amplification events previously uncovered in CRCs, such as CDK8, EGFR, CMYC, and ERBB2 gene amplifications, 18 the work uncovered gene amplification and overexpression of the IGF2 gene in about 10% to 15% of CRCs. 19,20 Gene deletions affecting well-defined tumor suppressor genes were confirmed in a subset of CRCs, such as those affecting APC, PTEN, or SMAD3. 19,20 In addition, frequent deletions affecting the FHIT presumptive tumor suppressor gene were seen in about 20% to 30% of CRCs. 19,20 Recurrent translocations activating two different genes encoding R-spondin protein family members that cooperate with Wnt ligands to activate canonical β-catenin-dependent Wnt signaling were seen in 5% to 10% of CRCs. 20
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


