Fig. 24.1
Nucleotide excision repair and the DNA damage response from radiation and chemicals, oncogene driven replication and virus infections. Adapted with modifications from ref. [33].
The DDR has been most extensively studied as a response to DNA double strand breaks (DSBs) , usually caused directly by X-rays [37]. Common assumptions assert that DNA double strand breaks activate the kinases ATM and Chk2, and replication stress (e.g., hydroxyurea, UV light) activates ATR and Chk1 [37–46]. ATM can also be activated by oxidative stress without necessarily involving DSBs [47]. The Chk1 and Chk2 kinases phosphorylate p53, cause nuclear translocation of the Cdc25 phosphatases, and increase expression of p21. Increased p21 results in G1/S, S, and G2/M cell cycle arrest, during which DNA repair can occur. Both ATM and ATR kinases are involved in the DSB and replication stress responses with considerable variation according to the damage and cell type [48]. At least 700 different protein substrates have been identified as targets for phosphorylation by these kinases [49]. There will be substantial differences in the structure of DNA and chromatin adjacent to DSBs compared with that resulting from replication arrest which lead to different signaling cascades and checkpoint activation [50–52]. The ATR pathway is essential for viability, ATR and Chk1 knockout strains are lethal [53]. ATM and Chk2 mutants are viable even though cells lacking these kinases display genomic instability and a predisposition to cancer [54]. The long history of sensitization of cells to UV damage by growth in caffeine [55] now appears to be explained at least in part by the sensitivity of the ATR-CHK1 pathway to caffeine [56, 57].
A major early signal of DNA damage and repair is the phosphorylation of the minor histone H2AX which occurs within minutes of DSB induction. H2AX is one of several nonallelic isoforms of histone H2A [58]. H2AX is ubiquitously distributed throughout the genome and has phosphorylation sites within the conserved C-terminal region at serine 139 and tyrosine 142 [59, 60]. Induction of DNA DSBs triggers H2AX phosphorylation (γH2AX) over megabases adjacent to DNA DSBs [60, 61]. After exposure to ionizing radiation, H2AX phosphorylation promotes chromatin condensation, which is detectable by immunofluorescence microscopy as nuclear γH2AX foci [60]. Initial recruitment of DNA repair and checkpoint factors, e.g., Mre11, Rad50, Nbs1, Brca1, and 53BP1, to DNA DSBs is not affected in H2AX knockout mice but repair and checkpoint factors subsequently fail to form foci, indicating that γH2AX functions to concentrate repair and checkpoint proteins in the vicinity of DNA lesions [62, 63].
The DDR from UV-induced photoproducts is more complex, and has distinct G1 and S phase responses. NER in G1, for example, excises UV photoproducts before onset of DNA replication and thereby eliminates mutagenic lesions completely [64]. NER is required during this phase for activation of the ATR-Chk1-p53 signal pathway [65]. Cells defective in the global branch of NER (XP-A through XP-G), but not the TCR branch (CSA, CSB), show reduced ATR-dependent signaling in G1 [65] and less γH2AX formation [66]. H2AX-knockout mice are cancer-prone and the knockout cells are sensitive to X-rays and etoposide that make DSBs [67], but show little sensitivity to UV light or cisplatin that do not produce direct DNA DSBs (Cleaver, unpublished observations). Therefore despite extensive H2AX phosphorylation after most forms of DNA damage, γH2Ax is only functionally important for DSBs but is only a biomarker for other forms of damage.
Damage that persists in S elicits a stronger ATR-Chk1-p53 response than in G1 [65], and contributes to recovery of DNA replication [68]. Stalled replication forks form long (kilobase) stretches of single-stranded DNA [44, 69–73]. These become coated with RPA that activates ATR through ATRIP [44]. The bypass polymerase Pol η is recruited through reversible ubiquitylation of PCNA [74–77]. A small fraction of arrested replication forks degrade to DSBs [78] or are resolved by nonhomologous or homologous recombination [79]. Replication fork breakage may be an active enzymatic process, and by analogy with DNA cross-links could involve either MUS81 or the NER nuclease XPF/ERCC1 [80]. UV-induced replication arrest results in large increases in γH2AX formation associated with coordination of the numerous replication and recombination factors associated with the arrested forks [66, 81, 82], but the absence of H2AX does not confer any increased UV sensitivity (Cleaver, unpublished observations). Resolution of the arrested replication forks also depends on p53, and cells lacking both p53 and Pol η are extremely sensitive to cell killing by UV light [83].
Resolution of arrested forks can result either in complete restoration of normal DNA replication, or large changes in genomic stability leading to mutagenesis or malignant transformation (Fig. 24.1).
24.3 Two Branches to Damage Removal
The main substrates for NER relevant to XP are the UV photoproducts between adjacent pyrimidines: the cyclobutane pyrimidine dimers and the [6-4] pyrimidinone dimers [84]. These are produced in DNA in skin over the whole UV wavelength range (UVA–UVB–UVC), although the most important wavelengths from solar exposure, from a practical standpoint, are UVB, approximately 280 to 320 nm [85]. The precise proportions of these photoproducts, and their pyrimidine constitution (T=T, C=C, T=C, and C=T) vary with wavelength, but in general they are the archetype large DNA adduct that is the main class of substrates for NER [86–89].
NER can remove DNA damage before DNA replication begins (Fig. 24.2), and consequently plays a major role in reducing the amount of damage that becomes fixed as mutations during replication [64, 90], but there is also evidence for a direct role in the UV damage response during the S phase, especially the ATR/Chk1 pathway [91]. The NER process has two main branches distinguished by the mechanisms used for initial recognition of DNA damage [92]: transcription coupled repair (TCR) by which actively transcribed genes are repaired more rapidly than the rest of the genome [93–97], and global genomic repair (GGR), by which non-transcribed regions of the genome are repaired [6]. Damage in transcribed genes is recognized through the arrest of RNA Pol II that is relieved through the action of two proteins, CSA and CSB. Damage in the non-transcribed or global regions of the genome is recognized by the binding of two heterodimeric XP proteins, XPE (DDB1/DDB2) and XPC/HR23B, which may act in concert to bind to damaged DNA [6]. TCR and GGR converge on a common pathway through which the damage is verified (XPA/RPA), the DNA is unwound by the XPB and XPD helicase components of the transcription factor TFIIH, cleaved by structure-specific nucleases (XPG, XPF/ERCC1), and the damage excised and replaced.
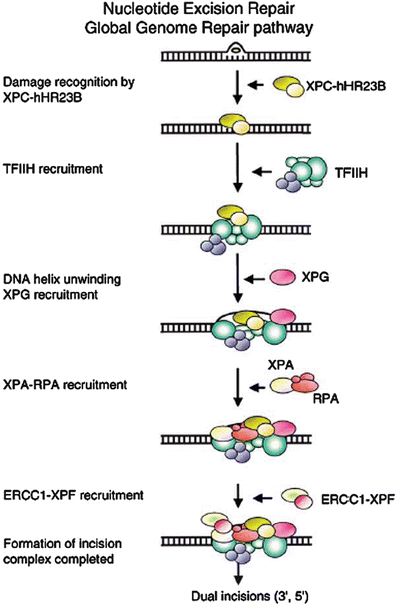
Fig. 24.2
Sequence of action of nucleotide excision repair (global genomic repair) on untranscribed DNA. Transcription coupled repair employs the arrest of RNA Pol II at DNA photoproducts as the damage recognition step, bypassing XPE and XPC, and entering the excision process at the TFIIH step. Reproduced from ref. [107] with permission from Elsevier.
The complete excision process on metabolically inactive DNA can be carried out in vitro with a minimal set of recombinant proteins: RPA, XPA, TFIIH (6 subunit core of XPB, XPD, p44, p34, p52, p62, and 3 subunit kinase CAK), XPC, hHR23B, XPG, ERCC1, and XPF [98–101]. Both TCR [102, 103] and GGR [104–106] are regulated by p53.
The sequence of action of the damage recognition, unwinding, and excision steps are interwoven rather than proceeding in a strictly sequential process [107]. The individual factors of NER associate independently on UV photoproducts without a necessary preassembly of a repairosome complex, although many of the components exist as heterodimers or within the larger 10-member TFIIH complex [100, 107–109]. The individual damage-binding proteins have relatively low affinity for damage as separate factors, but cooperatively XPA, XPC, and RPA, with cooperation from TFIIH, form a high specificity complex [110].
24.4 Global Genomic Repair
The initial damage recognition factors uniquely required for GGR are the XPC and XPE (DDB2) DNA binding proteins (Figs. 24.2 and 24.3), both of which function as heterodimers: DDB1/DDB2 and XPC/HR23B. Naturally occurring mutations in XPE (DDB2) have only a small effect on the levels of NER [111], whereas mutations in XPC cause a large reduction in NER and increased UV sensitivity. For this reason, XPC appears to be the major DNA damage recognition protein and XPE may play a lesser role, but this conclusion may be misleading. Mouse knockouts in Ddb2 [9] and virally induced downregulation of DDB1 [112] have a large influence on the excision process.
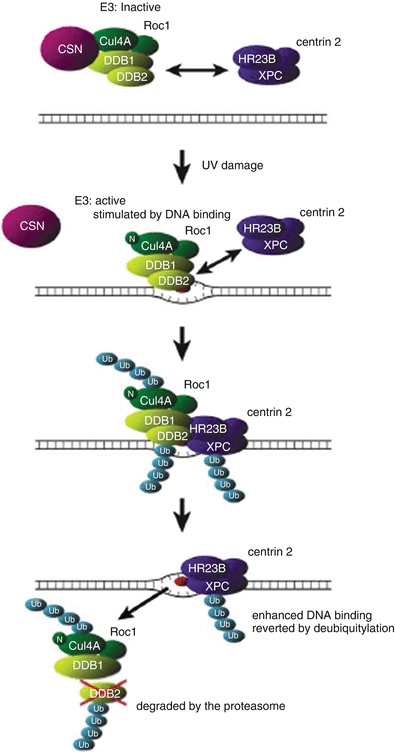
Fig. 24.3
Model for UV-Induced DDB1/2 dependent ubiquitylation of XPC. In unirradiated cells, DDB1/2-associated E3 is inactivated by its interaction with the COP9 signalosome (CSN). Thus, XPC is not ubiquitylated, despite its interaction with DDB1/2. Upon UV irradiation, UV-DDB translocates onto the damaged chromatin by binding to lesions, particularly [6-4] photoproducts. Its dissociation from the CSN and the neddylation of cullin 4A (indicated by “N”) activate E3. The binding of UV-DDB to the lesion further stimulates E3 activity. The activated DDB1/2-E3 then recruits XPC via protein–protein interaction, and XPC, DDB2, and cullin 4A are ubiquitylated at the lesion site. Polyubiquitylated UV-DDB1/2 loses its damaged DNA binding activity, whereas the DNA binding of XPC is potentiated by its ubiquitylation. This results in the displacement of UV-DDB by XPC on the lesion. The ubiquitylated DDB2 is degraded by the proteasome. The ubiquitylated form of XPC reverts to the unmodified form through deubiquitylation. Reproduced from ref. [285] with permission from Elsevier.
The XPE group includes patients who are mildly to moderately affected and whose cells carry out near normal levels of NER [113]. The XPE gene product is the smaller partner of a dimeric protein with subunits of 127 kDa (DDB1) and 48 kDa (DDB2) [104, 113–118]. Mutations that are diagnostic for XPE are located in the DDB2 subunit (see http://www.xpmutations.org for a summary of XP and CS mutations). DDB2 and p53 regulate each others’ expression [119, 120], with the consequence that XPE cells and those from mouse knockouts are resistant to p53-dependent UV induced apoptosis [9].
The DDB1/2 heterodimer is involved in the recognition of damaged DNA and has the capacity to bind to UV-damaged DNA with a chemically defined footprint [114, 121–124]. Binding appears specific for most cyclobutane and [6-4] photoproducts, except for the T[cis,syn]T photoproduct which is marginally discriminated from normal undamaged DNA [121]. DDB1 (p127) is present in excess over its partner, and is predominantly cytoplasmic but translocates to the nucleus after UV irradiation [125], a function that requires DDB2 [126], and has a large number of other functions apparently unrelated to NER.
XPC is the major damage binding protein that initiates NER [127]. XPC binds tightly with a cofactor HR23B, one of the two human homologs of the yeast Rad23 gene product, and both are located on chromosome 3p25, a region deleted frequently in human squamous cell carcinomas [128]. The XPC/HR23B complex [129, 130] binds to damage in non-transcribed DNA, acting to stabilize subsequent XPA/RPA binding to the damaged site, with a high affinity [92, 127]. XPC is displaced through ubiquitylation by the E3 ligase activity of DDB1/DDB2. XPC has sequence similarity to single strand binding proteins and DNA binding appears to be critically dependent on one tryptophan residue (Tryp690) [131]. The mechanism appears to involve binding to the undamaged strand depending on the relaxation of base-pairing in the damaged region, giving the recognition process great versatility and independence from the actual chemistry of the damaged site [131].
Residual repair in XP-C cells is confined to small transcriptionally active islands of the genome, where repair occurs at normal rates, surrounded by oceans of unrepaired transcriptionally silent DNA [132–135]. The XPC/HR23B complex is associated with CEN2, a protein that stabilizes XPC and which is also involved in centrosome duplication, suggesting a direct link between GGR and cell division [136]. The constitutive level of expression of XPC is controlled by p53, and expression can be induced by UV and ionizing radiation [137, 138].
24.5 Transcription-Coupled Repair
Pyrimidine dimers are excised more rapidly from actively transcribed genes, especially from the DNA strand used as the template for transcription [93–97]. A detailed base by base analysis of excision in the promoter and early region of the PGKI gene, using ligation-mediated PCR, revealed that excision is slow where binding proteins interact with the promoter but increases immediately after the ATG start site for transcription [142]. The initial damage recognition mechanism for TCR may be the stalled RNA Pol II, itself, although a potentially large number of gene products play a role in mediating the increased rate of repair in transcriptionally active genes [96, 97]. During transcription on a damaged template the pyrimidine dimer causes mis-incorporation of uridine into the messenger RNA, which blocks further transcription [143].
The arrest of RNA Pol II at sites of DNA damage during transcription elongation plays a central role for damage recognition in TCR [144]. The blocked RNA Pol II masks damaged sites and must be removed, or backed off, to alleviate the arrested transcription and provide access for repair enzymes. Recent evidence suggests that the CSB protein prevents Pol II back-tracking induced by TFIIS, to produce a stable stalled complex [143]. This complex provides time for the assembly of the NER excision complex and expansion of the transcription bubble by TFIIH. Excision then removes the damaged strand, the Pol II enzyme and the nascent transcript.
Cells from patients suffering from the neurodegenerative and sun-sensitive disease Cockayne syndrome are specifically defective in TCR [12, 145]. The excision of DNA photoproducts from total genomic DNA of CS cells is normal, but repair of transcriptionally active genes is reduced [145], and DNA and RNA synthesis fail to recover to normal levels after UV irradiation [146]. The neurodegenerative symptoms of CS could be a direct result of neuronal loss through apoptosis of non-dividing cells that experience natural oxidative damage in the brain. Two CS genes, CSA and CSB, are involved specifically in TCR. CSA contains WD-repeat motifs that are important for protein–protein interactions [147]. CSB contains an ATPase, helicase motifs, and a nucleotide binding domain, but only the latter is essential for TCR and CSB does not function as a helicase [148, 149] The CSB protein is associated in the RNA Pol II elongation complex with DNA and nascent RNA [150–152] and can actively wrap DNA [153]. The CSA protein is neither part of this complex, nor forms a stable complex with CSB [150], but is still required for TCR during transcription elongation [151, 154]. Cells lacking either CSA or CSB are unable to ubiquitinate the CTD of RNA pol II [155].
24.6 Damage Verification and Unwinding
After damage recognition through the specific GGR or TCR proteins, a process of damage verification appears to occur in which the region containing the damage is expanded into a single stranded bubble by the 3′-5′ (XPB) and 5′-3′ (XPD) helicases contained in the transcription factor TFIIH [109, 156]. The initial DNA binding proteins are replaced by another damage binding complex, XPA and RPA, that stabilizes the region and has a larger binding affinity [157, 158]. XPA binds to both the damaged and the undamaged strand, but RPA binds mainly to the undamaged strand [157]. XPA binds to the p34 subunit of RPA through its N-terminal region and to the p70 subunit through a central portion in exon IV; to ERCC1 through a small region in exon II; to DNA through its central region covering approximately exons III to V and a Zn finger in exon III; and TFIIH through its N-terminal region [159].
The transcription factor TFIIH plays a central role in unwinding the damaged region and downloading several of the other NER components. TFIIH can interact with all of the components of the core excision complex, individually, but its strongest binding under physiological conditions is with XPC and XPG [109]. Mutations in the XPB and XPD components can also affect the promoter opening function of TFIIH [160, 161]. The phenotypes of mutations in XPD are associated with three different clinical disorders: XP group D, TTD, or a rare combination of XP and CS. XP-B is similarly associated with a combined XP and CS disorder.
The XPB protein is required for both repair and basal transcription, whereas XPD is dispensable for basal transcription, which may explain why fewer surviving patients with mutations in XPB are reported than XPD [65]. Most TTD patients exhibiting UV sensitivity fall into the XP-D complementation group [30]. Human mutations that are relevant to the disease phenotype in the XPB and XPD genes are all mis-sense because loss-of-function mutations are lethal, although the severity can be influenced by gene dosage [30]. XPD is an essential protein for transcription and cell viability, and no patient can have two null alleles [30]. Several cases of XP-D and TTD patients have an allele in common, suggesting that the second alleles determine the clinical presentation, which is supported by studies in S. pombe showing the shared alleles behave as null mutations [162] and would play no direct role in the disease. The distribution of mutations within the XPD protein reveals that essentially all XP-D mutations fall within one of the conserved helicase domains [163]. This pattern indicates that these mutations can be expected to reduce the helicase activity of the protein [160]. In contrast, the TTD mutations usually fall outside of the helicase domains and show significant clustering at the C-terminus of the protein. The structure of the Archeal homolog of XPD has elucidated the functional consequences of many mutations by placing them within the structure of the protein [164, 165]. A mouse strain with a mutation that copies a human TTD mutation in XPD has similar symptoms to those seen in human TTD patients, together with an aging and cancer-prone phenotype [26].
TTD-specific mutations may subtly interfere with the ability of XPD to interact with its partner proteins within the TFIIH complex and thereby destabilize the complex [160, 166]. Evidence in support of this idea comes from the identification of a component of the transcription factor TFIIH corresponding to TTD-A patients that have no mutations in XPB or XPD but have low levels of TFIIH [167]. This component has a small size, only 8 kDa [168, 169] and is mutated in three patients with TTD, and appears to be required for maintenance of normal levels of TFIIH.
24.7 Excision Nucleases
The final stable complex that binds and remains at the site of DNA damage acts as an assembly point for the XPG and ERCC1/XPF nucleases that cut the damaged strand 3′ and 5′ to the damaged site. The excision process removes a 27–29 nucleotide oligonucleotide containing the photoproduct by cleavages 5 nucleotides on the 3′ side of the photoproduct, and 24 nucleotides on the 5′ side [170]. The structure-specific cleavage pattern is determined by binding of RPA to the unwound damaged site, and the excised fragment is close in size to the footprint of RPA on the DNA [171]. Slight variations in the precise sites of cleavage result in the removal of variable fragments between 27 and 29 nucleotides in size. The nucleases also function in the repair of DNA-DNA crosslinks [172], recombinational repair [173], chromatid exchange aberrations [174], and telomere maintenance [175, 176]. These nucleases a have sequence similarities to the FEN-1 class of nucleases and interact with PCNA and hence play a role in DNA replication [177, 178].
The 5′ cleavage occurs first and is carried out by a heterodimer of XPF and ERCC1 that incises DNA on the 5′ side of the damaged site [108, 123, 179, 180]. The XPF and ERCC1 genes may represent an ancient duplication and are unstable unless in a heterodimer complex [181]. The enzymatic activity resides in the XPF partner of the complex [182]. XPA serves as an anchor for the 5′ nuclease through binding to ERCC1 [183]. Patients with mutations in XPF are rare and most cases have been found in Japan with relatively mild symptoms [184]. Only one patient has been identified with mutations in ERCC1, which is associated with extremely severe symptoms [21]. In contrast knockout mice for both the XPF and ERCC1 genes are very severely affected and are neonatal lethal [31, 185, 186].
The XPG nuclease cleaves on the 3′ side of the damage and interacts with the XPC-hHR23B complex and TFIIH [109, 187]. The XPG nuclease has a complex range of activities, and mutations in XPG are associated with both XP and CS diseases [12, 188, 189]. XPG is an endonuclease in the FEN-1 family that is capable of strand-specific cleavage of a range of DNA substrates that may arise during DNA replication, repair, and recombination [173]. The enzyme binds to the single strand/double strand junction at the NER bubble but remains inactive until unwinding by TFIIH from the XPF/ERCC1 cleavage causes its activation. The protein not only cleaves the 3′ side of photoproducts during NER [190, 191], but it also resolves recombination intermediates by strand specific cleavage at double strand-single strand junctions [173, 192]. XPG is also a cofactor for thymine glycosylase through which it stimulates repair of oxidative DNA damage (thymine glycols, 8-oxoguanine, etc.) that may also be important in maintaining neural functions that fail in CS patients [191, 193, 194].
The XPG gene product interacts with RNA Pol II by interaction with the elongation factor TFIIS, independently of CSB and facilitates efficient transcription elongation [195], which provides an explanation for the complex symptoms of some XPG patients that show both XP and CS symptoms. The excised region is replaced by the action of a complex similar to that involved in normal DNA replication [101]. PCNA is loaded onto the DNA by the 5 subunit RFC complex, which then anchors the replicative polymerases Pol delta or Pol epsilon [196]. The cell cycle regulator p21 is upregulated after damage and slows S phase progression through binding to PCNA, but it appears to be specifically degraded during the repair polymerization. The final closure of the repaired site occurs with DNA ligase, most probably ligase I [196].
24.8 Nucleotide Excision Repair : Polymorphisms and Heterozygote Risk
The disease alleles of the NER genes represented in the human diseases XP, CS, TTD, and COFS represent high penetrance mutations that cause disease when homozygous [197]. There are in addition susceptibility alleles that may give rise to disease at lower penetrance according to life-style, diet, hormonal balance, and exposure to environmental chemicals, radiation, and infectious agents. A large number of alleles present at low frequency individually could combine to create variation in disease frequency in the overall population. Heterozygosity in XP genes has also been of concern, especially in parents of affected children.
Polymorphisms have been found in most of the XP genes and many disease associations have been claimed [198–202]. Many of these polymorphisms result in nonconservative amino acid changes that could have an impact on NER function and disease [203]. The reported associations between repair gene polymorphisms and disease suffer in many cases from problems of statistical power. A detailed reexamination suggests that as yet few of the reported associations are on sufficiently large data sets to be firmly established [199].
Heterozygosity in NER genes has not been clearly associated with significant disease among XP parents, although the evidence is largely anecdotal. Detection of small increases in skin cancer among XP parents is likely compromised by the small numbers of subjects, and low solar dose rate, especially if they share their children’s restricted sun exposure. In contrast several mouse strains with NER genes knocked out have shown increased cancer in the heterozygotes. These include the XPE (Ddb2) mouse [9], the Xpc mouse [10], and the Xpv (Pol eta) mouse [11]. These may represent increased ease of detection of an heterozygous UV sensitivity by the high UVB dose rates used in mouse cancer studies, as well as potential unique properties of the repair genes on mouse backgrounds as compared to human.
24.9 Replication of Large DNA Adducts: Class Y Bypass Polymerases
DNA photoproducts are blocks to the replicative DNA polymerases, alpha, delta, and epsilon that cannot accommodate large distortions such as DNA photoproducts or adducts in their active sites [204, 205]. Replicative bypass of these photoproducts is achieved instead by damage-specific polymerases with relaxed substrate specificity, now defined as class Y polymerases [206, 207]. Class Y polymerases have larger active sites that allow them to read through noninformative sequence information resulting from DNA damage [208]. As a consequence these polymerases have high error rates on the order of 1 % when assayed in vitro, and this property must be restricted in vivo to prevent catastrophic levels of mutation [209, 210].
Three class Y polymerases have been identified in the mammalian genome, Pol eta, iota, and kappa. The genes Pol eta and Pol iota are close homologs, unique to mammalian cells, and only a single Pol eta (yRad30) gene is found in yeast [206]. Pol iota has a poorer capacity for replication of UV damage and Pol kappa seems completely unable to replicate UV damage. Pol eta has a greater capacity for replicating T-containing photoproducts and Pol iota a preference for C-containing photoproducts. Pol eta can therefore insert adenines across from thymine-containing photoproducts accurately replicating a T=T pyrimidine dimer. Pol iota can insert guanines across from cytosine-containing photoproducts, resulting in accurate replication of a T=C pyrimidine dimer or [6-4] photoproduct.
Pol eta is regulated by several mechanisms that prevent extensive replication and restricts its subcellular location. Pol eta acts distributively, and extends nascent DNA chains by one or two bases across from the photoproducts, after which replication switches to Pol zeta or kappa [211]. Pol eta is uniformly distributed in the nucleus, and excluded from the replication fork until replication is stalled by UV damage, whereupon it relocates with other proteins in microscopically visible foci coincident with PCNA at the replication fork [74, 212]. Relocation requires specific sequence motifs at the C-terminal end of the protein for translocation and for binding to ubiquitylated PCNA [212, 213].
The error-free DNA damage replication pathway involving Pol eta and other polymerases is part of an overall DNA replication pathway for UV damage defined in yeast as the RAD 6 epistasis group [214]. Several of the components of this pathway are ubiquitinating enzymes that target PCNA, which functions as a major control point for the overall process [215]. PCNA is modified by UBC9 that is a small ubiquitin-related modifying enzyme (SUMO), and by Rad6 and the MMS2-UBC13 heterodimer that are ubiquitin-conjugating enzymes [215]. Multiple modifications compete for the same lysine (K164) residue on PCNA and redirect subsequent pathways through SUMO, monoubiquitination, and multiubiquitination [215]. Recruitment of Pol eta is regulated by reversible ubiquitylation of PCNA by the Rad6/Rad18 ligase and the USP1 deubiquitylating protease [77]. The monoubiquitylation favors recruitment of Pol eta, whereas Ub(K63) favors error-free bypass or recombination, and sumoylation favors normal DNA synthesis [76]. Delays in replication fork progression leads to autodegradation of USP1 and increased PCNA ubiquitylation which recruits Pol eta [77], as well as Pol iota and Pol kappa [216]. Pol eta is consequently recruited to the replication fork in response to more general cellular stresses than just its specific role in bypass of bulky UV DNA lesions.
Pol eta plays a large number of roles in situations where replication presents significant obstacles to normal replication polymerases as well as during important physiological processes. Pol eta is important, for example, during immunoglobulin hypermutation and contributes to generating the strand bias of A–T base pair substitution errors [217–225], and in homologous recombination during immunoglobulin conversion [226]. Pol eta also plays a role in the extension of invading strands in D-loops during homologous recombination to prevent replication fork degradation [227], although in contrast Pol eta deficiency in vivo increases homologous recombination [79]. Immunoglobulin hypermutation is also enhanced by other polymerases such as Pol iota [228] and Pol theta [229]. Deficiencies in nucleotide pools, such as caused by the inhibitor hydroxyurea, are also sufficient to recruit Pol eta to replication forks [82]. Pol eta activity is enhanced by interaction with other components of the damage response that accumulate at replication forks, such as the mismatch proteins MSH2-MSH6 [230], although mismatch repair generates a different mutation spectrum than Pol eta [223].
In the absence of the low fidelity polymerases, a complex set of overlapping intra-S phase checkpoints are activated that prevent gross chromosome rearrangements [81, 231, 232]. The extended regions of single stranded DNA remaining after the fork progresses without Pol eta increase signaling by the ATRIP-ATR kinase pathway and enhance S phase arrest [44]. Blocked replication forks are further stabilized in UV damaged cells by p53, and its inactivation by viral transformation allows fork breakage and DNA rearrangements to occur [74, 81, 83, 231, 233]. The actual frequency of fork breakage is low [78] and most arrested forks are resolved without breaks [68]. Central players in these rearrangements are the protein complex hMre11/hRad50/Nbs1 and Rad51 that are involved in nonhomologous and homologous recombination, DNA replication arrest, and telomere maintenance [81, 231, 232, 234].
24.10 Systemic Impacts of Nucleotide Excision Repair
24.10.1 Disorders of the Central Nervous System
Patients with mutations in the NER genes associated with TCR, both XP and CS, often exhibit progressive neurological abnormalities in addition to skin photosensitivity [235] and this is presumed to be caused by endogenous sources of DNA damage. Consistent with this attribution, mouse embryo fibroblasts derived from Csb −/− mice are sensitive to X-rays and oxidative damage [236–238], but in both species the sensitivity to oxidative damage is greater for mutations in CSB than CSA [236, 239].
The neurological symptoms include progressive mental deterioration, diminished deep tendon reflexes, and deafness sometimes beginning early in life [12]. Patients may have microcephaly, intellectual deterioration with loss of the ability to talk, and increasing spasticity with loss of ability to walk, leading to quadriparesis, in addition to dwarfism, and immature sexual development. The major histological feature is primary neuronal degeneration with dysmyelination, particularly in the cerebral cortex and cerebellum. CS patients show extensive cerebellar atrophy including a marked reduction of granule cells and the loss of Purkinje cells [13, 240].
The neurological symptoms of XP and CS patients have been ascribed to defective repair in the brain of endogenous oxidative damage that blocks transcription [191, 193, 237, 241–243]. The precise origin of oxidative damage is not known, but presumed to be mitochondrial leakage. Cerebellar granule cells are vulnerable to a variety of toxins that decrease glutathione levels and make the cells more vulnerable to cellular damage from reactive oxygen species [244, 245]. Purkinje cells are susceptible to ischemic death due to their reduced capacity to sequester glutamate and reduced ability to generate energy during anoxia [246]. Tissues that degenerate in XP and CS appear to be sensitive to oxygen levels, including the Purkinje cells, retina, and oligodendrocytes [247–250]. TUNEL staining and oxidative stress has been observed in cerebellar granule cells from clinical samples of CS and XP-A patients [251–254].
No consistent correlation has yet been established between sites of mutation and the severity of the clinical symptoms. Null mutations in the early region of the CSB gene have been found in a mildly UV sensitive patient with minimal neurodegenerative symptoms [14], and in quite severe patients [255, 256]. The presence of the PiggyBac transposon in intron 5 is flanked by CSB mutations and its contribution to the symptoms remains to be established [255, 257].
Mouse knockouts of XP and CS genes generally present a weaker neurological phenotype than the corresponding human disorder but greater cancer predisposition [8, 258, 259]. Xpa −/− mice, for example, have no neurological deficit, and only a slight reduction in lifespan [28, 29], in comparison to human XP-A patients who are usually severely affected. Cs-a and Cs-b mice, unlike the corresponding human CS disorder, are predisposed to UVB-induced and chemically induced skin cancer [259–262].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


