© Springer International Publishing Switzerland 2015
Janice L. Pasieka and James A. Lee (eds.)Surgical Endocrinopathies10.1007/978-3-319-13662-2_50VIPoma
(1)
Department of Surgical Oncology, St. Vincent’s Hospital and University of New South Wales, Suite 709 St Vincent’s Clinic, 438 Victoria St, 2010 Darlinghurst, NSW, Australia
Keywords
Multiple endocrine neoplasiaNeuroendocrine tumorsVasoactive intestinal polypeptideVIPomaIntroduction
VIPomas are rare neuroendocrine tumors usually occurring in the pancreas associated with the increased systemic release of vasoactive intestinal polypeptide (VIP) causing symptoms of high-volume watery diarrhea. Their management is complex involving surgery, medical therapies, interventional radiology, and nuclear medicine modalities.
Historical Features
The first report of a patient with VIPoma was likely by Priest and Alexander in 1957 [1]. They reported the case of a 56-year-old male who died from complications of refractory diarrhea, hypokalemia, and hyponatremia in association with a pancreatic neuroendocrine tumor that had recurred after resection. Verner and Morrison described this constellation of clinical features in more detail in 1958, and the syndrome produced by high levels of VIP released from these tumors subsequently bears their names [2]. They reported two male patients who died with similar clinical features of chronic high-volume watery diarrhea, hypokalemia, and hyponatremia, leading to their death from complications of dehydration and electrolyte disturbance. At autopsy, both were found to have pancreatic neuroendocrine tumors, and the authors postulated that a hormonally active substance produced by these tumors was responsible for the diarrhea and electrolyte disturbance. The hormone responsible was not identified until 1970 when Said and coworkers isolated a hormonally active peptide from porcine small intestinal cells and found that it increased splanchnic blood flow, naming it VIP [3]. An association between elevated levels of VIP and the Verner–Morrison syndrome was first shown by Bloom and coworkers in 1973 [4]. They found elevated levels of circulating VIP in patients with pancreatic neuroendocrine tumors and symptoms of the Verner–Morrison syndrome, and also showed staining for VIP within these tumors on immunohistochemistry. A direct association between VIP and the clinical symptoms of this syndrome was also shown by Kane and coworkers in 1983. They were able to induce a profuse watery diarrhea and electrolyte disturbance in human volunteers after infusion of porcine VIP [5].
Incidence and Risk Factors
VIPomas are a rare form of an already uncommon family of tumors, accounting for 1 % of all pancreatic neuroendocrine tumors [6]. It is estimated that they occur with an incidence of less than one case per million population per year [7, 8]. These tumors can occur at any age including childhood, with a mean age of between 40 and 50 years [9]. They occur with equal frequency in both males and females. They may be associated with other pancreatic neuroendocrine tumors, primary hyperparathyroidism and pituitary adenomas as part of the multiple endocrine neoplasia type 1 (MEN1) syndrome , occurring in 1–2 % of patients with this inherited genetic mutation [10, 11]. Eleven percent of patients with VIPoma managed at the Mayo Clinic had MEN1 [12].
Pathophysiology
VIP is a peptide hormone comprising 28 amino acids and normally functions as a neurotransmitter confined within neurones of the central nervous system, gastrointestinal tract, lung, and genitourinary tract. The plasma half-life of VIP in the systemic circulation is extremely short (less than 60 s), and for this reason plasma levels of this hormone are normally very low (less than 50–100 pg/ml) and do not elevate after eating [9]. VIP acts via binding to specific receptors to increase the secretion, smooth muscle contractility, and blood flow within the gastrointestinal tract, particularly the small intestine and pancreas.
The high circulating levels of VIP secreted by VIPomas produce symptoms of watery diarrhea, typically more than 6–8 l/day [9]. The diarrhea does not have features of malabsorption such as steatorrhea, and persists despite fasting. The loss of electrolytes in the stool, particularly potassium and bicarbonate, leads to hypokalemia and achlorhydria. For this reason, the syndrome has been previously referred to as WDHA (watery diarrhea, hypokalemia, and achlorhydria) syndrome and pancreatic cholera. Other symptoms include weight loss (occurring in 72 % of cases), generalized muscle weakness, abdominal pain (50 %), hyperglycemia (40 %), and cutaneous flushing (28 %) [12, 13].
Although VIP is the predominant hormonally active peptide produced by VIPomas, high levels of other hormones have also been documented in as many as 66 % patients with these tumors, including glucagon, pancreatic polypeptide, serotonin, calcitonin, somatostatin, insulin, gastrin, and growth-hormone-releasing hormone [12, 14].
Diagnosis and Imaging Studies
VIPomas are associated with very high circulating levels of VIP, with serum concentrations typically greater than 500 pg/ml (normal range less than 50–100) being diagnostic for these tumors [8, 15]. As for other neuroendocrine tumors, serum levels of chromogranin-A may also be elevated [16].
VIPomas occur most commonly in the pancreas, accounting for 90 % of cases. Within the pancreas, the tail of the gland is the most common location (50–75 %), followed by the body (8–22 %) and the head (11–30 %) [9, 12]. Rare sites of primary VIPomas include the duodenum, colon, bronchial tree, and liver. The adrenal glands and retroperitoneal paraganglia can also rarely be the site of these tumors particularly in children [8].
VIPomas typically form discrete masses within the pancreas. By the time of diagnosis, they have usually reached a size of more than 2 cm, with a mean size of 5 cm in one large series [9]. Local invasion into surrounding organs may be seen with larger tumors. Metastases are present in 60–80 % of cases at the time of diagnosis, most commonly to regional lymph nodes and to the liver (seen in up to 78 % of cases) [9, 12].
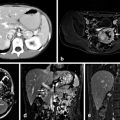
The location of the primary tumor is usually readily apparent on cross-sectional imaging using computed tomography or magnetic resonance [17]. As both primary VIPomas and their metastases are hypervascular, contrast enhancement aids in their imaging. Magnetic resonance has been recommended as superior to computed tomography scanning for the detection and staging of VIPomas [17].
VIPomas express receptors for somatostatin in 80–90 % of cases, and in this way nuclear medicine scanning using radiolabeled somatostatin analogs such as octreotide can detect 91 % of primary tumors and 75 % of metastatic lesions [9, 18]. More recently, positron emission tomography using the 68-gallium-labeled somatostatin analogs DOTATATE and DOTATOC have been used to enhance the sensitivity of these studies [18]. Small primary tumors of the pancreas may be best located by endoscopic ultrasound, a technique that also allows for fine-needle aspiration biopsy of lesions. This technique has been particularly recommended in patients with MEN1 syndrome who may have multiple small neuroendocrine tumors of the pancreas [17].
Management
The management of VIPomas is complex and multidisciplinary, involving surgical, medical, interventional radiology, and nuclear medicine modalities. Due to the rarity of these tumors, our understanding of their management is incomplete and evolving, based largely on expert opinion, anecdotal reports, and small case series. The complexity of the management of these rare tumors means that their treatment should be confined to specialist centers that can offer the full range of treatment options.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


