Mean age of onset (years)
Frequency (%)
Central nervous system (CNS)
Retinal hemangioblastomas
18
70
Craniospinal hemangioblastomas
29
60–90
Endolymphatic sac tumors
22–32
11–16
Visceral
Pheochromocytomas
30
10–20
Pancreatic lesion
36
17–75
Pancreatic neuroendocrine tumors (PNET)
35
20
Renal cell carcinoma or cysts
34
90–95
Epididymal cystadenoma
35
42
Broad ligament cystadenoma
–
–
Older estimates suggest the incidence of vHL to be 1:36,000 live births in an Anglo-Saxon population [3]. Substantial geographical variability and referral bias occur as patients live in proximity to their kindred and their care becomes increasingly concentrated at specialized centers. Thus, the true incidence is difficult to ascertain but a more recent estimate is 1:39,000–53,000 [2, 4]. The lifetime penetrance of vHL-associated disease in those with a vHL mutation is nearly 100 % [5]. Complications of renal cell carcinoma (RCC, 32 %) or CNS hemangioblastomas (53 %) continue to be the leading causes of vHL-related mortality [1, 2].
Characterization of the natural history of the disease as well as the genetic and molecular abnormalities in vHL has resulted in substantial improvement in the management of these patients. In addition, it has led to the ability to inform genetic counseling and provide prognostic determinants based on disease subtype. The knowledge gained about the disease has become the basis for current screening and surveillance guidelines as well as impacted surgical decision making. A study of survival rates of multiple genetic neoplastic disorders comparing before and after the intensification of standardized screening demonstrates significant progress in the management of vHL. The life expectancy for vHL prior to 1990 and the establishment of a genetic registrar was 52.5 years and had the lowest expectancy of the diseases studied. A survival benefit of 16.3 years was seen in those patients diagnosed after 1990 increasing the life expectancy to 68.8 years [6]. Screening of asymptomatic vHL patients and at-risk family members identifies disease-related lesions at an earlier stage and correlates to higher cure rates for malignant lesions as well as increased options for organ-sparing interventions. Guidelines are established and are readily available to patients and providers through the vHL Family Alliance at www.vhl.org. These guidelines are summarized in Table 2 and will be revisited throughout this chapter.
Age | Test | Comments |
|---|---|---|
0 | Physical examination Routine newborn hearing screening | Pediatrician assess for neurological disturbances or ophthalmic abnormalities (nystagmus, strabismus, white pupil, etc.) |
1–4 | Indirect ophthalmoscope (annual) Physical examination (annual) | Ophthalmologist performed eye/retinal examination Regular assessments with pediatrician that includes the above as well as blood pressure, vision, and hearing abnormalities |
5–15 | Same as above plus: Fractionated metanephrines or 24-h urine test (annual) Abdominal ultrasound (annual after 8 years of age) Complete audiology assessment (every 2–3 years) | If biochemical abnormalities or concerning findings on ultrasound are found, an abdominal MRI or MIBG scan is indicated If symptoms of audiovestibular disturbance are found, audiology screening frequency should be annually MRI with and without contrast of internal auditory canals in case of repeated ear infections |
16 + | Same as above plus: MRI of abdomen wwo contrast (every 2 years) MRI of the brain and entire neuroaxis (every 2 years) | Neuroaxis includes cervical, thoracic, and lumbar spine with thin slices through posterior fossa/temporal bone |
Pregnancy | Retinal screening (periodic) Fractionated metanephrines or 24-h urine test (per trimester) MRI of brain and spine (4th month of gestation) | Hemangioblastomas are known to have accelerated growth during pregnancy Important to ensure the absence of a pheochromocytoma during pregnancy and prior to delivery |
The crucial importance of coordinated screening and intervention for a disease that, by definition, involves multiple organ systems makes the case that vHL patients should be referred to dedicated vHL care centers with an experienced and appropriately resourced multidisciplinary team. Specialized care centers are essential for optimum surveillance, coordinated operative and nonoperative care, and ancillary service support. The endocrine surgeon plays an important role in the management of vHL but must be placed appropriately within the context of whole-patient disease management system.
Disease Subtypes
vHL patients can be classified into clinical subtypes (Table 3) with major subtypes based on the risk of developing a pheochromocytoma . Type 1 patients may develop all other tumor types associated with vHL; however, they have a significantly reduced risk of pheochromocytoma. Type I accounts for 80–93 % of the vHL population. Type 2 patients have a high lifetime risk of a pheochromocytoma and represent the other 7–20 % of vHL patients [2]. Type 2 patients can be further subdivided into groups (A, B, or C) on the basis of the likelihood of developing RCC . Within the type 2A subgroup, there is a predominance of pheochromocytomas with a low risk of RCC versus in type 2B where there is a high risk of RCC, retinal, and cerebral disease in addition to a significant risk of developing a pheochromocytoma. The only manifestation of vHL in type 2C patients is pheochromocytoma. Despite these general classifications, due to variable penetrance in patients with the same mutation, screening and management guidelines are not currently tailored based on subtype classification.
Table 3
vHL disease subtypes
Type 1 | Reduced risk of pheochromocytoma |
Types 2 | Increased risk of pheochromocytoma |
Type 2A | Increased risk of pheochromocytoma + low risk of RCC |
Type 2B | Increased risk of pheochromocytoma + high risk of RCC |
Type 2C | Pheochromocytoma only |
Diagnosis, Molecular Biology, and Genetics
vHL exhibits autosomal dominant inheritance with > 95 % penetrance of disease specific manifestation by age 65. The involved gene in vHL maps to the short arm of chromosome 3, specifically 3p25-p26 [7]. There also exists a prominent modifier of the vHL gene by CCND1 on 11q13.3. The vHL gene is a tumor suppressor gene and, as such, the germline mutation in one allele is not adequate to result in emergence of tumors. Per Knudson’s two-hit hypothesis, a second mutation must occur with the normal vHL allele resulting in “loss of heterozygosity” that causes complete loss of the gene’s normal downstream products and effects [8]. This “second hit” results in loss of tumor suppressor function and subsequent tumor development.
The vHL gene is fundamental in the response to hypoxia and serves as a cellular oxygen sensor. It is important in angiogenesis, erythropoiesis, and cell cycle regulation [9, 10]. The protein product (pVHL) is a component of an E3 ubiquitin ligase complex, which normally mediates degradation of the hypoxia-inducible factor (HIF) transcriptional complex via poly-ubiquitination in oxygen-replete cells. In contrast, its function is suppressed in hypoxic cells, which allows for activation of the HIF target genes associated with angiogenesis, growth, and mitogenic factors, extracellular matrix control, and microtubule regulation, as well enhanced glucose uptake [9–11]. In vHL, regulation of HIF is lost. The affected tissues behave as if in a chronically oxygen-depleted environment with an uncontrolled growth stimulus. The most pronounced impact is seen in highly vascularized areas such as the CNS, retina, medulla, kidneys, and pancreas [12].
Genetic testing has revealed over 100 germline mutations associated with vHL among known kindreds. Frequent mutations include R238Q and R238W, which is associated with a 62 % incidence of pheochromocytoma , as well R167Q and R167W, which are associated with developing both pheochromocytoma and renal cell carcinoma. Generally, type I disease is more associated with deletions, nonsense, and frameshift mutations, whereas type II disease is associated with missense mutations. While these examples demonstrate that there is a correlation between specific mutation and disease manifestations, the same mutation in different individuals may have different clinical manifestations. Due to the overall poor correlation between genotype and phenotype, the specific mutation cannot be used to tailor screening recommendations, or treatment strategies, at this time [13, 14].
If the patient has a previously described mutation in the vHL gene, genetic testing can confirm the diagnosis with near 100 % specificity [15]. In the absence of an already confirmed mutation status, the established clinical criteria may be used to establish diagnosis. In the setting of a positive family history, the patient in question must present with any one major disease-related lesion, which include a CNS hemangioblastoma, retinal hemangioblastomas , pheochromocytoma, or RCC . In the setting of no family history, the diagnosis requires two vHL-associated lesions, which can include two or more CNS hemangioblastomas or one CNS hemangioblastoma and one visceral tumor [2].
Specific Manifestations
We will individually address the organ-specific manifestations of vHL, progressing generally from cephalad to caudad. We will address greatest attention to those neoplasms that are of greatest clinical impact to the patient, as well as those of particular interest to the endocrine surgeon.
Cerebral and Spinal Hemangioblastoma
Hemangioblastomas of the CNS are capillary-rich, circumscribed, benign neoplasms. Although benign, due to their location in the neuraxis, they can cause major morbidity and mortality due to mass effect of the lesion (as seen in the patient in Fig. 1), associated cysts, or from hemorrhage. CNS hemangioblastomas are the most prevalent neoplasms in vHL, developing in 60–90 % of patients [16]. These present at a younger average age than sporadic hemangioblastomas (mean of 29 years in vHL) and more often are multifocal [17]. Both sporadic and vHL-associated hemangioblastomas typically are infratentorial (Figs. 1 and 2). A prospective evaluation of 250 vHL patients with CNS hemangioblastomas demonstrated a median of eight lesions per patient, with 1 % supratentorial, 6 % in the brainstem, 45 % in the cerebellum, and the remaining 48 % caudal to that (spinal cord, cauda equina, and nerve roots). These patients developed a median of 0.3 new lesions/patient-year [16]. Although also infratentorial, a larger proportion (> 80 %) of sporadic CNS hemangioblastomas are cerebellar [17].
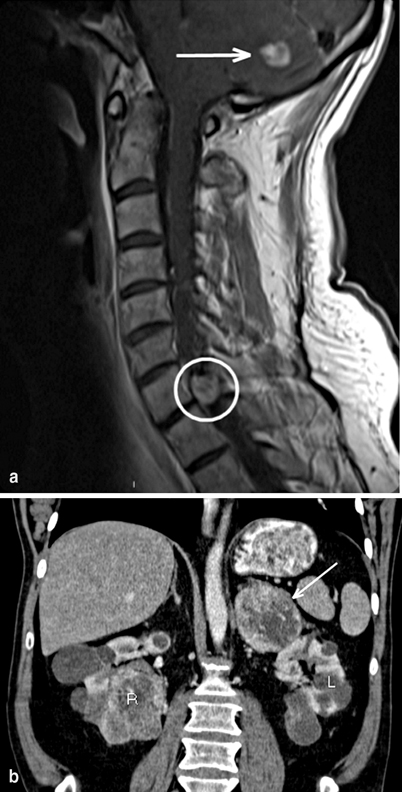
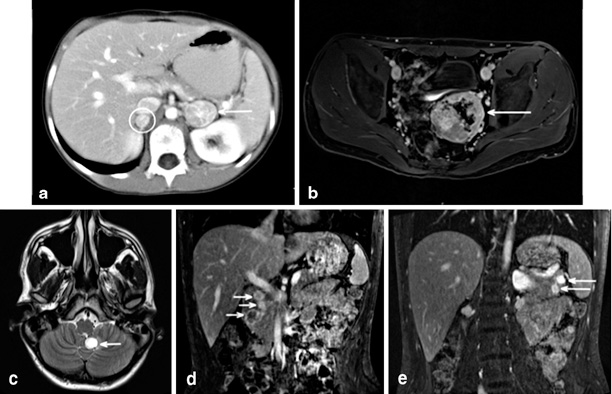
Fig. 1
Radiographic findings all from a single 47-year-old male without prior diagnosis of von Hippel–Lindau (vHL), but a suggestive family history, who presented with chief complaint of headache. Imaging demonstrates two cerebellar hemangioblastomas, one causing obstructive hydrocephalus ( arrow in image a), as well as spinal hemangioblastomas (including at C7 in circle in image a). Recent-onset hypertension and clinical suspicion based on multiple hemangioblastomas prompted biochemical testing and abdominal imaging. Computerized tomography (CT) demonstrated 7 cm left adrenal lesion consistent with pheochromocytoma ( arrow in image b), an unremarkable right adrenal gland and multiple bilateral renal cysts and masses consistent with RCC (see R and L in image b). Twenty-four-hour urine collection demonstrates norepinephrine 557 (15–80 μg/24 h), epinephrine < 5 (0.5–20 μ/24 h), and normetanephrines + metanephrines 5.4 (0.17–0.70 mg/24 h)
Fig. 2
Radiographic findings all from a single patient from a known vHL kindred. He underwent bilateral total adrenalectomy at age 10 for bilateral pheochromocytoma ( circle and arrow image a). At that time his biochemical workup included 24- h urine with epinephrine < 5 (< 25); NorEpinephrine 552 (< 100); NorMet + Met 3.5. He was lost to formal follow up until re-presenting at age 20 following a school entrance physical. Imaging at that time demonstrates a large pelvic paraganglioma (image b), cerebellar and spinal hemangioblastomas (image c), three head of pancreas PNETs (image d), and two tail of pancreas PNETs. Biochemical evaluation revealed no evidence of functional PNET, and urine normetanephrines 1.2 nMol/L (< 0.9), urine metaneprhines < 0.20 nMol/L
Other than presentation with a symptomatic lesion, identification of CNS hemangioblastoma occurs by imaging. The majority of lesions are clinically asymptomatic and there are no available screening laboratory studies. CT is not an adequate test for this entity and contrast-enhanced magnetic resonance imaging (MRI) is the preferred modality. Current guidelines for screening in vHL patients are for ≤ 1.5 T MRI scans with and without contrast to include the brain and whole spine with thin cuts through the posterior fossa. These are to start by age 16 and be performed every 2 years. This interval is then shortened in those with demonstrated lesions.
The majority of these lesions are asymptomatic and do not require intervention. However, due to the presence of multiple lesions per patient, many vHL patients will harbor at least one symptomatic lesion. At final evaluation in a prospective series, while only 6 % of all detected lesions had associated symptoms requiring intervention, they were distributed across one-third of the patient population [16]. The rate of growth of vHL-associated CNS hemangioblastoma varies with the anatomic location of the tumor, with more rapid progression for those in the brainstem and cerebellum [16]. These lesions may develop cysts in some cases (12 %) and the presence of a cystic component is associated with faster growth and a greater likelihood of developing symptoms [16].
It is not universally agreed when to intervene on these lesions, but most believe that they should be closely observed radiographically, and correlated with history or physical exam findings for specifically associated neurologic symptoms, and that onset of symptoms should prompt treatment of the causative lesion [16]. While there are some clinical variables that are associated with lesion progression [16], these are neither sensitive nor specific. Therefore, radiographic and clinical follow up is mandatory. When treatment of CNS hemangioblastoma is deemed necessary, surgical resection is the primary modality. Due to the morbidity and difficulty in surgical access to some of these lesions, stereotactic radiosurgery has also been employed. Although this therapy was well tolerated, stereotactic radiosurgery demonstrated diminished ability to control lesions over time. Therefore it is recommended that stereotactic radiosurgery be reserved for those symptomatic lesions not amenable to surgical therapy [18]. Although the vascular nature of the lesions raised hope that antiangiogenic therapy would be effective, there does not currently exist a demonstrated effective option for medical therapy.
While the presence of multiple CNS hemangioblastomas should very strongly raise the concern for vHL, an apparently sporadic CNS hemangioblastoma may be the index presentation of a previously unknown germline vHL mutation. In one series, this occurred in 4 % of patients otherwise thought to have sporadic CNS hemangioblastomas, all of whom presented at an age less than 40 years. A further 5 % of these patients who tested negative for germline mutation subsequently developed another vHL-related lesion, which may have been the result of mosaicism [19]. Given the importance of appropriate screening for other vHL-associated lesions, this is of clear importance for those patients in whom a germline vHL mutation is detected. For this reason, it is reasonable to submit all patients with retinal or CNS hemangioblastoma for vHL testing, even if these initially are believed to be sporadic lesions.
Retinal Capillary Hemangioblastomas
Hemangioblastomas also occur in the eyes of vHL patients and can be complicated by vision loss. These are frequently the earliest manifestation of vHL, and most vHL patients do not develop new retinal hemangioblastomas late in life. However, cumulatively, 70 % of vHL patients have retinal hemangioblastomas by age 60 and they frequently are bilateral. Compared to patients with sporadic retinal hemangioblastomas, they occur at an earlier age (18 vs. 36 years) and more frequently are multifocal and bilateral (about 50 %) [16, 20].
Retinal hemangioblastomas are not initially clinically apparent and are only detected by dilated ophthalmologic exam. Due to subretinal edema, traction, and hemorrhage, these lesions can result in vision loss. Monitoring and therapy aim to prevent or minimize vision loss. Treatment of most of these lesions is laser photocoagulation. There is not universal agreement and some recommend treating asymptomatic lesions while others recommend that small asymptomatic lesions may be closely monitored for growth or symptoms [16, 21]. As with CNS hemangioblastoma, investigations into antiangiogenesis therapies have demonstrated some potential role, but are not yet adequately studied.
Although not universally agreed upon, it is reasonable to offer vHL testing to patients with otherwise apparently sporadic retinal hemangioblastoma . In the presence of a known vHL germline mutation, screening for retinal hemangioblastoma is recommended annually starting at age 1 and in the hands of an ophthalmologist with expertise in these lesions.
Endolymphatic Sac Tumors of the Middle Ear
Tumors of the endolymphatic sac (ELST) are an overgrowth of the papillary epithelium of the endolymphatic system located within the middle ear. These tumors are characterized as highly vascular and locally aggressive. While rare in the general population, they are detected in 11–16 % of vHL patients, 22 with an age at onset of 22–32 years [23]. vHL is the only condition associated with bilateral ELSTs [23]. Patients may present with acute-onset hearing loss, tinnitus, pain, vertigo, facial paresis, and aural fullness [22–24]. The mechanism for the audiovestibular dysfunction’s sudden onset is hypothesized to be acute intralabyrinthine hemorrhage. However a more gradual decline in hearing secondary to otic capsule invasion and/or hydrops formation also is reported [22, 23]. Regardless of mechanism, hearing loss usually is permanent.
Screening for ELSTs begins at birth with the routine newborn hearing screening. Periodic assessments of hearing should be performed by the child’s pediatrician. Starting at age 5, a formal audiology examination should be completed every 2–3 years, and more frequently if symptoms arise. The vHL Alliance also recommends an MRI with contrast of the internal auditory canal with thin cuts in cases of recurrent ear infections every 2 years after age 16 which is concurrent with the exam for CNS hemangioblastoma [25]. Indications for surgery include patients with radiologic evidence of a tumor on computed tomography (CT) or MRI that either have preserved hearing, or for deaf patients with persistent neurological symptoms associated with an ELST [22]. Surgical resection does not improve hearing but has been shown to prevent further damage, and improve other associated neurological symptoms. Of vHL patients who complain of audiovestibular symptoms, 60 % do not have findings on imaging and surgical intervention of these patients remains controversial [22].
Pheochromocytoma and Paraglanglioma
Pheochromocytoma has a reported incidence of 0.1–0.5 % in the general population which correlates to 2–8 people per million per year. The “rule of 10s” previously supported the belief that approximately 10 % of pheochromocytomas occur in the setting of heritable familial syndromes. This has proven to be an underestimate as more syndromes have been recognized and testing has become more frequent [26]. Some estimates suggest that 24–30 % of sporadic pheochromocytomas occur within familial syndromes including vHL, neurofibromatosis 1, multiple endocrine neoplasias type 2, and succinate dehydrogenase mutations [27, 28]. More recently, this statistic has been contested citing referral and catchment area bias, and revised estimates are between 11.6 and 19 %. Within vHL patients, the rate of pheochromocytomas is 10–20 % [14]. However, the true risk of developing a pheochromocytoma depends on the type of vHL, varies among kindreds, and ranges from 0 % to upward of 57 %.
The demographics and manifestations of pheochromocytoma in vHL differ significantly compared to patients with sporadic disease, as well as when compared to other familial pheochromocytomas . The mean age of presentation is 30 years but has been reported prior to the age of 10 [14]. Pheochromocytomas in vHL patients are less likely to present with the classic signs and symptoms (palpitations, headache, diaphoresis, hypertension, tachycardia, pallor, and nausea) than sporadic pheochromocytoma. Over one-third of pheochromocytoma in vHL are clinically asymptomatic. Most pheochromocytomas in vHL are within the adrenal gland and up to 50 % of vHL patients have bilateral intra-adrenal disease. However, extra-adrenal paragangliomas also are more prevalent compared to sporadic disease, with 12 % of vHL patients having extra-adrenal disease, and some patients having bilateral intra-adrenal as well as extra-adrenal disease (Fig. 2). Paragangliomas can arise in the glomus jugulare, carotid body, and the periaortic tissues. Pheochromocytoma in vHL is less likely to have malignant transformation than pheochromocytoma in sporadic disease, with a rate of 3.3 %.
Patients with a diagnosis of vHL or at-risk family members with a known carrier status for a germline mutation should undergo routine screening for pheochromocytoma regardless of their disease subtype. Screening for pheochromocytoma should include both biochemical and radiographic testing. Biochemical screening is recommended to begin annually no later than 5 years of age with some studies suggesting initiating screening even earlier [14]. Serum chromogranin A is a nonspecific marker with poor sensitivity and is not a standard part of screening. Plasma-free metanephrines and normetanephrines are used as the first-line screening tool in vHL as in sporadic disease with a sensitivity of 97–100 % and specificity of 85–89 %. As with testing in sporadic disease, attention must be given to the medication list and testing environment to limit the rate of false-positive results. If the results from the serum testing are equivocal, the patient should conduct a 24-h urine collection for catecholamines. When performed correctly, urine biochemical studies have a sensitivity and specificity of 98 and 98 %, respectively, for pheochromocytoma; however, they are cumbersome for the patient to collect and are prone to errors in collection.
Regardless of the method of testing, pheochromocytoma in vHL has a predictable biochemical signature, with elevations in norepinephrine and normetanephrine, without elevations in epinephrine and metanephrine levels (as demonstrated by both patients in Figs. 1 and 2). In contrast, sporadic disease and other familial syndromes such as multiple endocrine neoplasia (MEN) have concurrent elevations of epinephrine and metanephrine [29]. The reason for primary production of norepinephrine in vHL patients is the relative lack of phenylethanolamine-N-methyltransferase (PNMT) responsible for the conversion of norepinephrine to epinephrine found exclusively within the adrenal medulla. vHL patients also express less tyrosine hydroxylase (the rate-limiting enzyme in catecholamine synthesis) than do patients with MEN2.
The age for initiating radiographic imaging for intra-abdominal manifestations of vHL is no later than 8 years of age. Family history and physical examination may suggest the need for earlier imaging as well as biochemical testing on a case-by-case basis and left to the discretion of the individual’s physician. The imaging modality of choice for screening of at-risk (known mutation carriers with no previous intra-abdominal lesions and negative biochemical studies) and asymptomatic established vHL patients is abdominal ultrasonography. If biochemical abnormalities are found, the most appropriate follow-up imaging study is an abdominal MRI in an effort to minimize the cumulative ionizing radiation exposure over a lifetime of repeated imaging studies. This is in contrast with sporadic disease, which can be evaluated with a dedicated adrenal protocol CT of the abdomen and pelvis with intravenous and oral contrast if there are no allergic or renal contraindications to intravenous contrast. CT and MRI have been shown to be equally efficacious.
In cases where CT/MRI are negative but there is high clinical suspicion or abnormalities in biochemical studies, a metaiodobenzylguanidine (MIBG) scan can be performed for localization with sensitivity of 95 % [30]. Recently, using CT/positron emission tomography (PET) with 18F-Dopa to localize pheochromocytomas has been studied. Small studies have shown a nearly 100 % sensitivity for pheochromocytomas. Unfortunately, the availability and acceptance of the test have been inhibited in part due to high cost and difficulty of synthesizing the 18F-Dopa [31]. All imaging modalities will have a lower sensitivity for small-volume disease.
Some image-detected pheochromocytomas in vHL do not result in elevated biochemical levels of catecholamines and there is no consensus on intervention or observation in that scenario. While some will intervene on any detected pheochromocytoma, others advocate withholding surgical intervention until the 24-h urine catecholamine excretion is double the upper limit of normal, or to remove nonfunctional pheochromocytoma if the tumor has grown to greater than 3.5 cm in diameter, the patient is to undergo an abdominal operation for another pathology, or if the patient desires to become pregnant or becomes pregnant [32].
Traditionally, the treatment of intra-adrenal pheochromocytoma is ipsilateral adrenalectomy, which classically was performed using an open technique by either a transabdominal or a posterior approach. With the advent of laparoscopy, today’s standard is a laparoscopic approach by either the lateral transabdominal or the posterior retroperitoneal approach, depending on the preference and expertise of the surgeon and center. There also are experiences with single port, robotically assisted, and single port robotic approaches to this procedure through both the transabdominal and retroperitoneal approaches. It is the opinion of these authors that there is at most marginal superiority of any one of these variations on minimally invasive adrenalectomy, beyond the significant benefit that resulted with dissemination of minimally invasive adrenalectomy techniques in general. Regarding the transabdominal versus the retroperitoneal approach, we select the approach best suited to the patient’s anatomy and prior operative history.
Traditionally, adrenalectomy is performed as a complete extirpation of the gland in question. Either sequential or simultaneous total adrenalectomy renders the patient surgically an-adrenal, and thus completely dependent on exogenous steroids, with associated quality-of-life impairment and risk for Addisonian crisis. This approach mandates thorough patient counseling and education. More recently, multiple large case series have demonstrated partial adrenalectomy (i.e., cortical-sparing adrenalectomy) via open, lateral or posterior laparoscopy, and robotic-assisted approaches to be safe and efficacious [33–35]. This involves removing the target lesion from the adrenal gland and leaving behind the grossly and radiographically normal portion of the gland. The main adrenal vein may be spared or sacrificed depending on the location of the tumor. The venae comitantes running with the arterial supply are adequate to maintain drainage even if the main adrenal vein is sacrificed, and as little as 10 % of a viable single adrenal gland has been shown to maintain adequate function to handle stress without requiring exogenous steroids [36]. Cortical-sparing adrenalectomy has been validated in the pediatric population with hereditary forms of pheochromocytoma [37]. The ability to leave a viable cortical remnant allows patients the opportunity to retain adrenocortical function, without dependence on exogenous steroids, while achieving comparable biochemical responses. Rates of Addisonian crises and long-term steroid replacement are significantly lower compared to complete adrenalectomy [33, 36, 38, 39]. As expected, recurrence rates are higher and time to recurrence is shorter with partial adrenalectomy. Outcomes data suggest these rates to be within an acceptable range and successfully treated with surveillance screening and additional surgery. The National Cancer Institute published their experience on hereditary pheochromocytoma as having a 6 % recurrence rate, which has been reproduced at other high-patient volume centers [38, 40]. In patients with vHL, the recurrence rate after partial adrenalectomy has been reported to be from 10 to 20 % after at least a 5-year follow up [39, 41]. The recent acceptance of cortical-sparing adrenalectomies makes it impossible to comment on the rates of patients that subsequently develop malignant pheochromocytoma and underscores the ongoing need for lifelong surveillance after surgical resection. In instances where additional pheochromocytomas develop, the benefit of adrenal-sparing surgery lies in delaying the development of adrenal insufficiency and subsequent lifetime duration of exposure to exogenous steroid. Therefore, the decision to perform cortical-sparing adrenalectomy versus total adrenalectomy must be made with consideration of the patient’s willingness and ability to tolerate adrenal insufficiency versus recurrence of disease with subsequent reoperation. The ultimate success of both approaches demands ongoing patient follow up.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


