1
Introduction
It is now well established that cardiovascular disease (CVD) represents the leading cause of mortality in both type 1 and type 2 diabetes and that the incidence of CV events and the mortality rates are significantly higher than in the general population. Subjects with type 2 diabetes (T2DM) have a two-to-fourfold higher CVD risk and threefold increased risk of CV mortality compared to those without diabetes [ ]. In type 1 diabetes (T1DM), the relative CVD risk is four-to-tenfold higher, and CVD events occur 10–15 years earlier than in nondiabetic subjects [ ].
Vascular calcification, the pathological deposition of calcium-phosphate (Ca-Pi) crystals within the arterial wall and the aortic valve, is considered a hallmark of both atherosclerosis and calcific aortic valve disease (CAVD). Indeed, calcium deposition is observed in advanced atherosclerotic plaques and the presence of calcified nodules represent the pathological peculiarity of stenotic aortic valve leaflets. Medial calcification, the accumulation of Ca-Pi crystals in the media layer of arterial vessels, is strongly associated with aging of the CV system but also represents a well-known pathological characteristic in the arteries of diabetic patients (the so-called “Monckeberg’s sclerosis”). Although the clinical relevance of vascular calcification is still controversial, several clinical and preclinical evidence clearly shows that this phenomenon is more frequent and accelerated in all the CV districts of diabetic patients.
Hyperglycemia is a relevant risk factor for vascular calcification in both diabetic and prediabetic subjects. Indeed, the prevalence of coronary artery calcification (CAC), marker of subclinical coronary atherosclerosis, is almost double in diabetic compared to nondiabetic subjects. Nevertheless, higher levels of HbA1c (glycated hemoglobin A1c) are associated with an increased prevalence and progression of CAC, even in metabolically healthy individuals [ , ]. In the Multi-Ethnic Study of Atherosclerosis (MESA), subjects with metabolic syndrome and diabetes had a greater CAC incidence and absolute progression compared with those without these conditions (adjustive relative risk 1.8; 95% CI, 1.4–2.2) [ ]. Moreover, the higher prevalence of CAC in diabetic individuals is associated with higher mortality if compared to nondiabetic subjects [ ]. Even in patients with type 1 diabetes, elevated CAC scores are strongly associated with higher absolute risk of CVD events and major adverse cardiac event (MACE) [ ].
Diabetes is also a relevant predictor of accelerated aortic valve calcification. Indeed, diabetes was correlated to a greater risk of developing CAVD in both the MESA study (odd ratio 2.06, 95% CI, 1.39–3.06) and the CANHEART study (hazard ratio 1.49; 95% CI, 1.44–1.54) [ ]. The MESA study also found a linear relationship between the presence of metabolic syndrome and the development of incident aortic valve calcification, measured by CT scan. Nevertheless, the highest odd ratio for incident valve calcification was detected in diabetic patients [ ]. The presence of diabetes, even at mild or well-controlled stage, is also a risk factor for aortic valve degeneration after valve replacement [ ].
2
Types and sites of vascular calcification
Cardiovascular calcification is due to the pathological deposition of calcium-phosphate crystals in the CV system, mainly the aortic valve leaflets, the aorta, and its major branches. Different kinds of vascular calcification can be classified based on the anatomical site and formation: passive or dystrophic calcification, caused by excessive Ca-Pi deposition on soft tissues, and three types of active calcification that are distinguished according to the location (aortic valve, intima layer, or media layer of the vessel wall), as shown in Fig. 4.1 . These three forms of active calcification can occur independently of each other, but often coincide and overlap. Nevertheless, the majority of subjects with vascular calcification have a predominant arterial calcification type; among them, diabetic patients have a higher prevalence of predominant medial calcification compared to intimal calcification [ ].
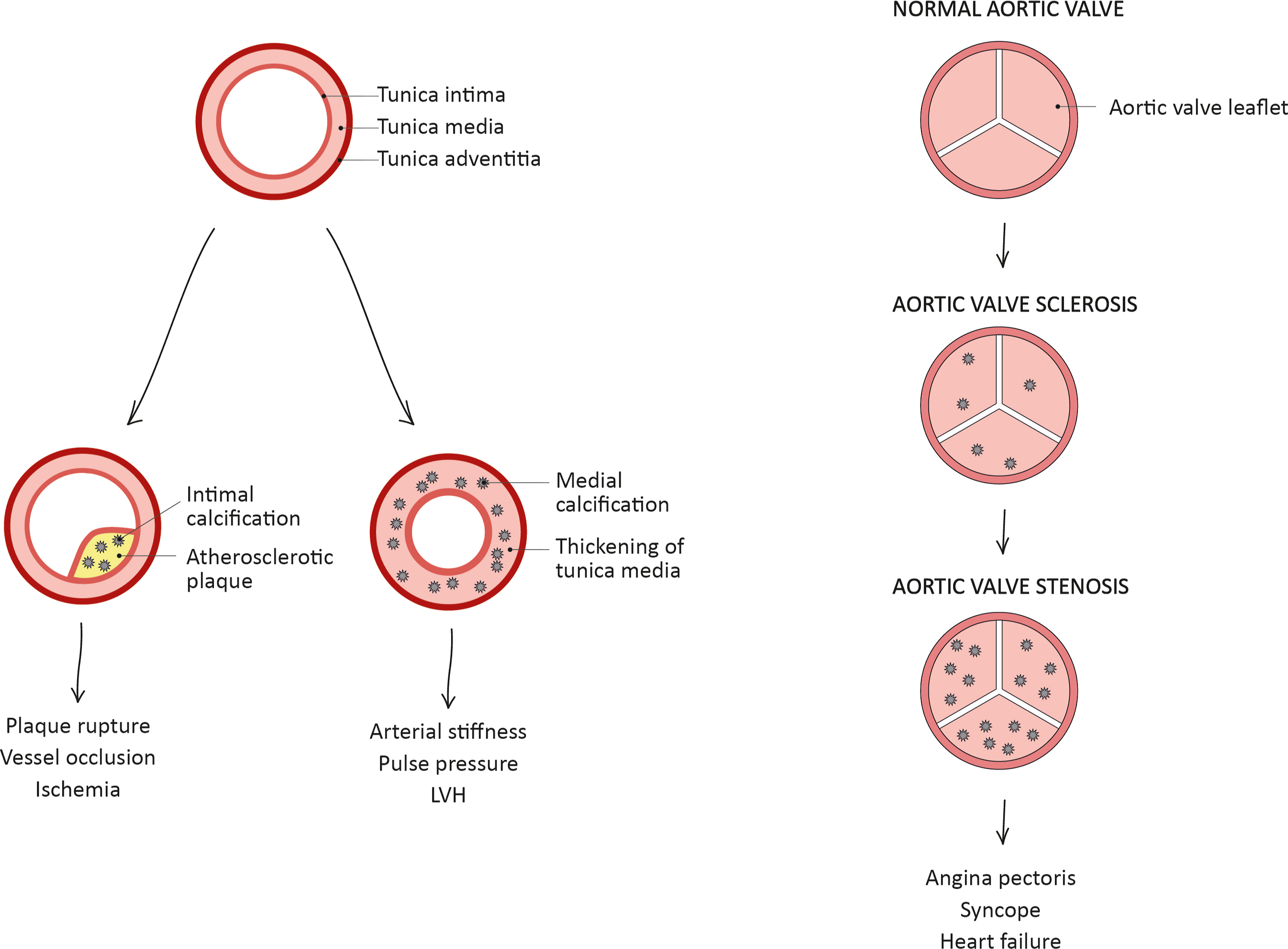
The three types of active calcification develop with different pathophysiological processes: intimal calcification is inflammation-driven and is typically located at the base of atherosclerotic plaques, adjacent to the medial layer. Valve and medial calcifications are mainly mediated by the differentiation of resident cells (interstitial valve cells [VICs] and vascular smooth muscle cells [VSMCs] respectively) toward an osteoblast-like phenotype with the upregulation of osteogenic markers, similarly to skeletal bone formation (see below for detailed mechanisms). Intimal and valve calcifications are characterized by lipid deposition, macrophage activation, and infiltration of inflammatory cells, while medial calcification typically occurs in the absence of these factors [ ].
Intimal calcification can be further classified based on the size of calcification: microcalcification (0.5–15 μm in diameter), punctuate calcification (15 μm-1 mm in diameter), fragmented calcification (≥1 mm in diameter), and sheet calcification (>1 quadrant of the vessel or >3 mm in diameter). In the intima layer, the initial microcalcifications can agglomerate to form at first punctuate areas of calcification, then fragmented calcification, also involving the surrounding extracellular matrix (ECM), and the necrotic core. Intimal calcification further diffuses with the progression of atherosclerotic plaque, reaching areas of sheet calcifications; this type of calcification incorporates VSMCs and ECM independently from the necrotic core [ ].
3
Risk factors for vascular calcification
The different types of active calcification are induced by separate risk factors: intimal calcification is mainly dependent on the traditional risk factors for CVD, including high blood pressure, elevated cholesterol levels, smoking, and inflammation. Medial calcification is independent on atherosclerotic process and may occur even in subjects with no traditional risk factors for CVD. Arterial medial calcification is frequently observed in chronic conditions, including diabetes, chronic kidney disease (CKD), and hypertension; however, the mechanisms leading to this type of calcification are still not completely understood [ ].
Compared to nondiabetic individuals, diabetic patients are characterized by a higher expression of osteogenic markers, including type I collagen, alkaline phosphatase (ALP), and an increased calcification in the medial layer of the vessels [ ]. Even the aortic valve of diabetic patients shows higher grade of mineralization and increased expression of early osteogenic markers, including ALP and Runx2 [ ]. Moreover, serum from T2DM patients has higher calcification potential (CP) than that from metabolically healthy subjects, suggesting that diabetes increases the risk of developing vascular calcification by conferring CP to serum [ ]. Taken together, these findings indicate that vascular calcification is accelerated in the arterial wall and aortic valve of subjects with diabetes. In line with this view, diabetes enhances vascular calcification in animal models of atherosclerosis ( Ldlr −/− mice ) [ ] and valve calcification ( Ldlr −/− ApoB 100/100 mice) [ ].
Hyperglycemia plays a crucial role in the pathophysiology of both intimal and medial vascular calcification. Indeed, hyperglycemia contributes to the development and progression of intimal calcification, mainly by increasing oxidative stress, promoting endothelial dysfunction and activating proatherogenic pathways [ ]. Hyperglycemia is also actively involved in the transdifferentiation of VSMCs toward an osteoblast-like phenotype. Indeed, the exposure of VSMCs to high glucose medium enhances ECM calcification and increases the expression of several bone-related markers, including ALP, osteocalcin (OCN), Runx2, Sox9, and bone morphogenetic protein-2 (BMP-2), if compared to euglycemic conditions [ ].
Lipid abnormalities, such as dyslipidemia and obesity, are common in people with type 2 diabetes, increasing the risk of developing vascular inflammation and calcification. Increasing preclinical evidence supports the direct involvement of dyslipidemia in vascular calcification. Indeed, in the aorta of high fat diet (HFD)-fed mice, the expression of several inflammatory and procalcific markers (such as TLR4, PKC, TGFβ, BMP-2, and NF-κB) is strongly higher than in normal diet fed-mice, favoring vascular inflammation and VSMCs differentiation. An HFD also enhances the serum levels of AGEs and the expression of their receptor RAGE in the aortic tissue, promoting the progression of vascular calcification through the activation of AGEs-RAGE signaling pathway [ ]. On a cellular level, LDL in the oxidized state (oxLDL) induces the osteogenic differentiation of VSMCs via p38 MAPK signaling pathway, promoting vascular calcification; conversely, HDL can prevent this process [ , ].
Hypertension is associated with remodeling of vascular tissues, including the arterial wall and the aortic valve. In particular, high systemic pressure leads to deposition of ECM components (such as collagen, fibronectin, and proteoglycans), increased activity of metalloproteinase (MMPs), activation of TGFβ signaling, and generation of elastin degradation products. Taken together, these factors create a favorable environment for the initiation and propagation of calcium deposition in the arteries [ ]. In the aortic valve, hypertension increases the mechanical forces in the aortic side of the leaflets and induces an overexpression of markers involved in inflammation, ECM remodeling (including MMPs, collagen), and calcification (such as osteopontin), favoring the progression of valvular calcification [ ].
It is well known that the renin-angiotensin-aldosterone system (RAAS) plays a major pathogenic role for the differentiation of resident cells in the arterial wall. In particular, angiotensin II (Ang II) can promote osteogenic differentiation of VSMCs through RANKL activation [ ]. Moreover, the treatment with an Ang II type I receptor blocker, upregulated in calcified arteries, is able to prevent vascular calcification [ ]. Hypertension is also a relevant risk factor for diabetes-associated vascular complications and a common comorbidity in T2DM patients. A meta-analysis of seven clinical studies showed that hypertension followed by diabetes are the most relevant risk factors for the prediction of CAC presence and extent, suggesting that both conditions result in a common arterial wall damage and consequent vascular calcification [ ]. The close relationship between diabetes and hypertension is due to common risk factors, including endothelial dysfunction, atherosclerosis, dyslipidemia, and obesity and similar pathogenic mechanisms, such as RAAS upregulation, oxidative stress, inflammation, and activation of immune system [ ].
Inflammation and oxidative stress are among the most relevant promoters of vascular calcification, mainly in the first stages of the disease. High oxidative stress induces the oxidation of several compounds directly involved in this process: for instance, both HDL and LDL once oxidized in the subendothelium become pro-osteogenic for VSMCs [ , ]. Toll like receptors (TLRs), which are known to be mediators of inflammatory responses, are overexpressed within atherosclerotic lesions. Recent in vitro evidence demonstrated that TLR4 is actively involved in the oxLDL-induced phenotypic change of VSMCs toward an osteoblast-like phenotype through the activation of NF-κB/ceramide pathway [ ]. Inflammation is also a potent inducer of arterial intimal calcification. Indeed, macrophages infiltration and local inflammation in the intima layer observed at early stages of atherosclerotic plaque induce the initial calcium deposition as microcalcification nuclei. In addition, inflammatory cytokines released from immune cells, including interleukin 6 (IL-6), interleukin 1β (IL-1β), and tumor necrosis factor-α (TNF-α), induce the transdifferentiation of VSMCs toward an osteoblast-like phenotype, promoting further mineralization within plaque lesions and creating a vicious circle of inflammation and calcification [ ]. These cytokines play a relevant role also for the osteogenic differentiation of VICs within the aortic valve. In particular, the binding of TNF-α to its receptor on VICs’ surface activates osteogenic pathways, including ERK1/2 and p38 MAPK. Moreover, the in vitro treatment of VICs with IL-6 has been shown to increase the expression of genes involved in their osteogenic differentiation, including BMP-2 and Runx2 [ ]. Furthermore, diabetes increases the valvular expression of NF-κB and in loco inflammation within the aortic valve in patients with concomitant CAVD, contributing to promote disease progression [ ]. It appears that all these inflammatory processes are amplified in diabetic subjects and are likely to be involved in ectopic calcification correlated to diabetic vascular complications.
4
Molecular mechanisms of vascular calcification
4.1
Osteogenic differentiation
The medial layer of the arterial vessels is mainly composed of elastin-rich ECM and contractile VSMCs, which normally express smooth muscle cell markers, such as smooth muscle-myosin heavy chain (SM-MHC), smooth muscle α-actin (αSMA), and smooth muscle 22α (SM-22α). As anticipated above, in response to external stimuli, VSMCs can undergo differentiation toward an osteoblast-like profile, characterized by the downregulation of smooth muscle contractile proteins and the overexpression of osteogenic markers, including ALP, Runx2, BMPs, and OCN [ ]. Several signaling pathways are activated in pathological conditions; among them, the canonical Wnt/β-catenin is particularly involved in osteogenic transition of VSMCs ( Fig. 4.2 ). Once differentiated, VSMCs secrete Ca-Pi-rich extracellular vesicles, which induce further ECM mineralization. These vesicles contain ALP, the osteogenic enzyme that hydrolyzes pyrophosphate (PPi), increasing the levels of inorganic phosphate [ ]. Furthermore, VSMCs can undergo apoptosis in response to proinflammatory cytokines, oxLDL, and mechanical injury, releasing apoptotic bodies that act as scaffolds for ECM mineralization. The apoptosis of VSMCs also contributes to arterial medial degeneration and increased vulnerability of atherosclerotic plaques [ ].
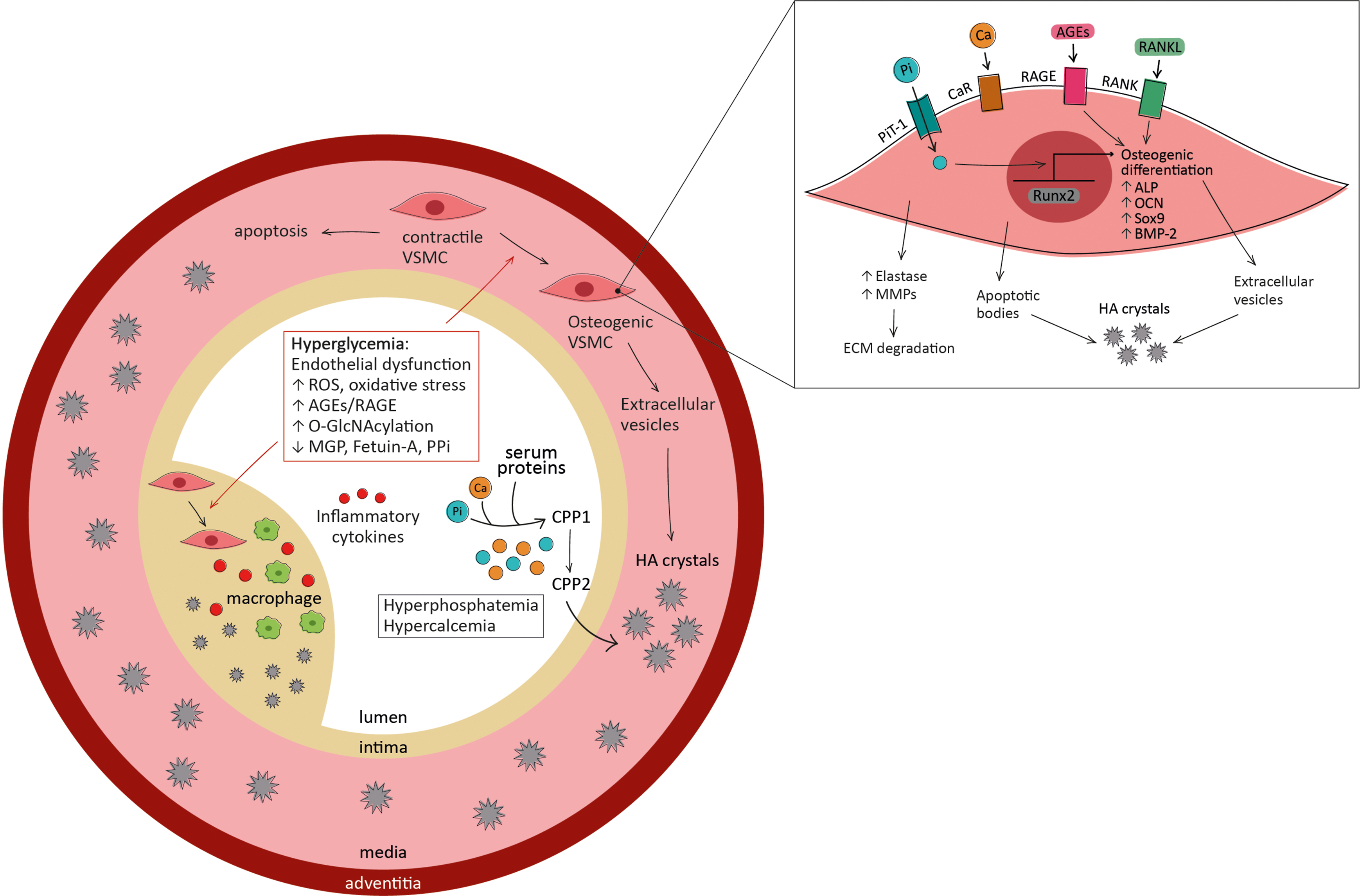
VSMCs are also one of the main inducers of intimal calcification, which is typically located adjacent to the internal elastic lamina: indeed, apoptosis of VSMCs promotes early calcification in the intima and osteogenic VSMCs co-localize with Ca-Pi deposits in atherosclerotic lesions. Although the phenotypic switch of VSMCs associated with calcification is likely to be the same in both intimal and medial layers, the factors driving this transdifferentiation are different. In medial calcification, the main drivers of VSMCs phenotypic transition are the loss of calcification inhibitors (such as matrix Gla protein (MGP), fetuin-A, and PPi), cellular senescence, uremia, and elevate Ca-Pi levels. Conversely, VSMCs phenotypic change in intimal calcification is induced by the activation of inflammatory pathways. Oxidative stress, VSMCs apoptosis, and mechanical forces are promoters of VSMCs osteogenic differentiation in both intimal and medial calcification [ ].
As mentioned above, elevated levels of glucose play a major role in osteogenic differentiation of VSMCs. Chronic hyperglycemia enhances the O-GlcNAcylation of several proteins including AKT, which is activated after glycosylation and, in turn, promotes osteogenic differentiation of VSMCs in vitro [ ]. Moreover, in high glucose conditions, the O-GlcNAcylation of several transcription factors (including Sp1, NF-κB, and Runx2) activates the proliferation, inflammatory responses, and osteogenic transition in VSMCs [ ].
4.2
Calcium and phosphate
Passive mechanisms of vascular calcification are mediated by increased concentration of calcium and phosphate. Elevated Ca-Pi levels induce hydroxyapatite nucleation and crystal growth, promoting ECM mineralization in the arterial wall. In the absence of mineralization inhibitors (such as MGP, fetuin-A, and PPi), the process exacerbates in vascular calcification.
In VSMCs, high intracellular calcium leads to cell apoptosis and enhances the production, release, and mineral nucleation of extracellular vesicles. The accumulation of mineralized vesicles and apoptotic bodies in the ECM facilitates crystal growth and induces further mineralization. In addition to promoting vascular mineralization, high calcium levels also drive phenotypic changes in VSMCs. The exact mechanism through which calcium influences cell osteogenic differentiation is unknown, but it seems likely that it involves the calcium-sensing receptor (CaR) on VSMCs’ surface: indeed, its depletion is associated with increased calcification [ ].
Hyperphosphatemia is also highly associated with vascular calcification through the induction of several different mechanisms. Hyperphosphatemia is a very common condition in CKD patients, due to renal function impairment and the consequently reduced excretion of phosphate. In the medial layer of the arterial wall, high phosphate levels induce ECM degradation, VSMCs apoptosis and osteogenic differentiation, and crystal deposition in the ECM [ ]. In particular, hyperphosphatemia promotes the self-assembly of amorphous Ca-Pi and serum proteins into calciprotein particles (CPPs) and their maturation from primary amorphous CPP1 to secondary crystalline CPP2 [ ]. In the healthy population, phosphate levels are also correlated to the presence and progression of CAC, a marker of intimal calcification [ ].
The phosphate-induced osteogenic differentiation of VSMCs and mineralization of ECM are mediated by PiT-1, the type III sodium-dependent phosphate transporter responsible for intracellular phosphate uptake. In response to high extracellular phosphate levels, PiT-1 acts as phosphate sensor and promotes ERK1/2 phosphorylation, driving osteogenic phenotype change [ ]. Moreover, in hyperphosphatemic conditions, PiT-1 is upregulated and can in turn promote calcium entry in VSMCs, further inducing vascular calcification [ ].
Interestingly, hyperglycemia and hyperphosphatemia have combined effects in promoting calcification, phenotypic transition, and PiT-1 expression in isolated VSMCs [ ]. These in vitro findings suggest that both glucose and phosphate levels should be monitored in patients with diabetic kidney disease.
4.3
Other regulators of vascular calcification
The balance between promoters and inhibitors of calcification is fundamental to maintain normal vessel physiology. Under pathological conditions, this balance is modified, with an increase in promoters and a loss of inhibitors, resulting in unregulated induction of osteogenesis [ ]. One of the most relevant calcification inhibitors is MGP, expressed by VSMCs, chondrocytes, fibroblasts, and endothelial cells in the arterial vessel wall. MGP can be activated by vitamin K through the γ-carboxylation of Glu residues; the carboxylated form (cGMP) binds to calcium ions, inhibiting the formation of hydroxyapatite crystals and therefore avoiding ectopic mineralization [ ]. Active cMGP can also interact with BMP-2, reducing its osteogenic activity in VSMCs [ ]. The protective role of cMGP in preventing vascular calcification is supported by preclinical evidence: indeed, the silencing of MGP gene induces arterial calcification in a mouse model [ ]. In patients with T2DM, the plasma levels of inactive MGP are independently and positively associated to peripheral arterial calcification and arterial stiffness, marker of medial calcification, suggesting a protective role of cGMP in diabetic vascular calcification [ , ].
Another relevant pathway involved in vascular calcification is the osteoprotegerin (OPG)/receptor activator of nuclear factor kappa B (RANK)/RANK ligand (RANKL) signaling axis. RANKL promotes the transdifferentiation of VSMCs toward an osteogenic phenotype in a RANK-dependent way [ ]. OPG, a soluble decoy receptor for RANKL, can counteract this process by preventing RANK/RANKL interaction and subsequent downstream cascade.
Among the pathways involved in diabetic vascular calcification, the activation of RAGE signaling pathway also plays a major role. In the CV system, RAGE is expressed on the surface of several cell types and can be activated by a wide range of ligands, such as AGEs and many endogenous molecules of the S100 calgranulin family. In diabetes, the expression of RAGE and its ligands (including AGEs, S100A8, S100A9, S100A12) is particularly increased [ ]. Recent preclinical findings demonstrate that, in high-glucose conditions, the activation of S100A9-RAGE axis induces the release of procalcific extracellular vesicles from pro-inflammatory macrophages, while diabetic mice lacking S100A9 develop less arterial calcification. These findings suggest a direct role of the activation of RAGE axis in macrophages in promoting diabetic vascular calcification, probably via Nrf2 and NF-κB pathways [ ].
The activation of RAGE also exerts a direct effect in phenotypic transition of VSMCs: indeed, AGEs production can induce calcium deposition and osteogenic differentiation of VSMCs in a RAGE-dependent manner [ ]. Moreover, RAGE activation in VSMCs reduces the expression of SM-MHC and SM-22α, proteins that are usually expressed by contractile VSMCs and are decreased after their osteogenic differentiation [ ]. Increasing evidence suggests that AGEs-induced vascular calcification is mediated by oxidative stress [ ]: indeed, the activation of AGEs-RAGE signaling in VSMCs induces NF-κB activation and ROS production via NAD(P)H oxidase [ ]. AGEs-mediated ROS generation also promotes VSMCs apoptosis, creating scaffolds for further vascular calcification [ ]. Moreover, the AGEs-RAGE downstream signaling promotes the activation of specific isoforms of protein kinase C (PKC), which can induce several signaling pathways, including the osteogenic p38 MAPK and ERK1/2 [ , ].
4.4
Aortic valve calcification
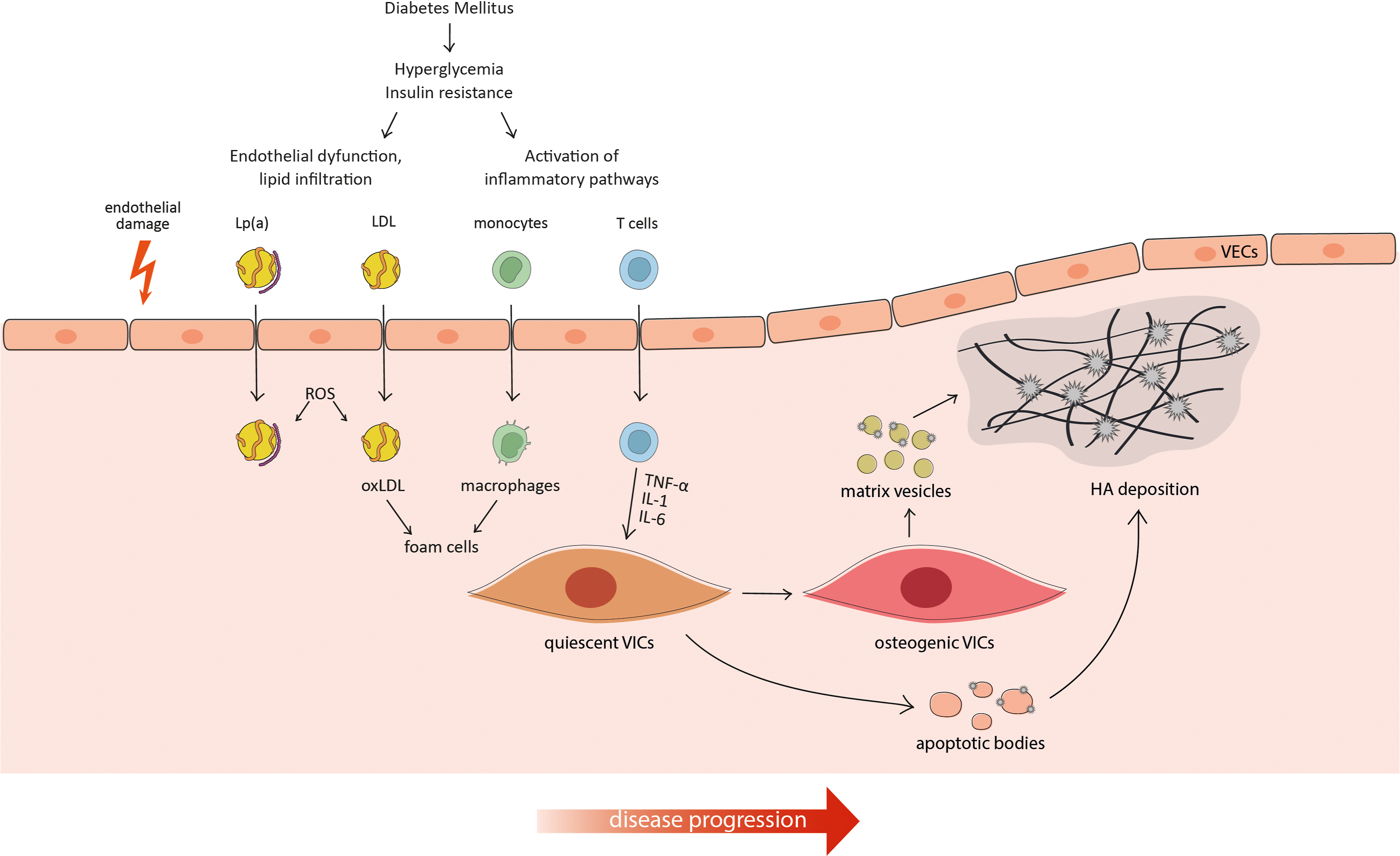
Calcification in the aortic valve leaflets is correlated to poor prognosis and adverse CV events, including heart failure, syncope, and angina pectoris. Therefore, once CAVD is symptomatic, the only available treatment to prevent mortality is the surgical or transcatheter replacement of the valve. Most of the pathogenetic mechanisms and risk factors described above are also involved in the progression of aortic valve calcification, including hypertension, dyslipidemia, and metabolic syndrome [ ]. As for intimal and medial calcification, aortic valve calcification is mainly driven by cell differentiation to an osteogenic phenotype. In this case, aortic VICs undergo differentiation in response to oxidative stress, modified lipids, and inflammation, acquiring a phenotype that over-expresses the main osteogenic markers. The phenotypic switch can be induced by several signaling pathways, including Wnt/β-catenin,RANK/RANKL, and BMPs. Once differentiated, VICs can also promote ECM mineralization through the release of Ca-Pi rich extracellular vesicles, which progressively aggregate and act as scaffolds for crystals deposition in the ECM [ ] ( Fig. 4.3 ).
Related posts:
 Overview on chronic complications of diabetes mellitus
Overview on chronic complications of diabetes mellitus
 Molecular mechanisms involved in hyperglycemia-induced cellular damage
Molecular mechanisms involved in hyperglycemia-induced cellular damage
 Epidemiology of chronic complications of diabetes: A global perspective
Epidemiology of chronic complications of diabetes: A global perspective
 Pathogenesis of diabetic atherosclerotic cardiovascular disease
REGULATION OF THE GROWTH HORMONE RECEPTOR AND BINDING PROTEIN
Pathogenesis of diabetic atherosclerotic cardiovascular disease
REGULATION OF THE GROWTH HORMONE RECEPTOR AND BINDING PROTEIN
 Glucose Prediction in Insulin-Dependent Diabetes
Glucose Prediction in Insulin-Dependent Diabetes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


