(1)
Department of Surgery, Dar A lAlafia Medical Company, Qatif, Saudi Arabia
3.1 Introduction
The normal hemoglobin is made up of two parts (Fig. 3.1):
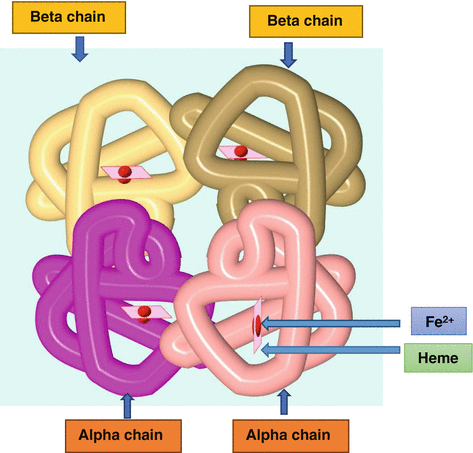
Fig. 3.1
Diagrammatic representation of the normal hemoglobin which is made up of heme and globin (2 alpha chains and 2 beta chains)
Heme
Globin
The heme is composed of iron plus a protoporphyrin molecule.
The globin part is made up of four polypeptide chains, two alpha chains, and two beta chains.
Each of the two alpha chains is made up of 141 amino acids.
Each of the two beta chains is made up of 146 amino acids.
The two alpha chains are derived from genes in the alpha gene cluster on chromosome 16.
The two beta chains are derived from genes in the beta gene cluster on chromosome 11.
3.2 Normal Hemoglobins
Hemoglobin A
This is the normal hemoglobin that exists after birth.
Hemoglobin A is a tetramer made up of two alpha chains and two beta chains (a2b2).
Hemoglobin A2
This is a minor component of the hemoglobin found in red blood cells after birth.
Hemoglobin A2 consists of two alpha chains and two delta chains (a2d2).
Hemoglobin A2 generally comprises less than 3 % of the total red cell hemoglobin.
Hemoglobin F
Hemoglobin F is the predominant hemoglobin during fetal development.
Hemoglobin F is a tetramer made up of two alpha chains and two gamma chains (a2g2).
The genes for hemoglobin F and hemoglobin A are closely related, existing in the same gene cluster on chromosome 11.
Hemoglobin F production falls dramatically after birth, although some people continue to produce small amounts of hemoglobin F for their entire lives.
In healthy adults, 95 % of the Hb is Hb A (α2β2) with small amounts (<3.5 %) of Hb A2 (α2δ2) and HbF (α2γ2) present.

Normal Hemoglobins
Hemoglobin A: two alpha chains and two gamma chains (a2g2)
Hemoglobin A2: two alpha chains and two delta chains (a2d2)
Hemoglobin F: two alpha chains and two gamma chains (a2g2)
Normal Hemoglobins in Adults
Hb A: 95–98 %
Hb A2: 2–3 %
HbF: 0.8–2 %
Hb S: 0 %
Hb C: 0 %
Homozygous HBS
HbA: absent unless transfused
HbS: over 50 %
HbF: up to 10 %. Higher for infants
HbA2: 2–4 %
Sickle Cell Trait
HbA: typically 50–65 %
HbS: typically 35–45 %
HbA2: normal (1.5–4 %)
HbF: higher for infants
3.3 Abnormal Hemoglobins
There are hundreds of hemoglobin variants that involve genes both from the alpha and beta gene clusters.
Hemoglobin variants
Hb S
Hb C
Hb E
Hb D-Punjab
Hb O-Arab
Hb G-Philadelphia
Hb Constant Spring
Hb Hasharon
Hb Korle-Bu
Hb Lepore
Hb M
Hb Kansas
Hemoglobin S:
This is the predominant hemoglobin in people with sickle cell disease.
This results from a single change of one amino acid, where glutamic acid at the sixth position of the 146 amino acids of the beta chain of hemoglobin is replaced by valine.
The molecule structure is a2bS 2.
People who have one sickle mutant gene and one normal beta gene have sickle cell trait which is considered benign.
Hemoglobin C:
Hemoglobin C results from a mutation in the beta globin gene.
This results from a single change of one amino acid, where glutamic acid at the sixth position of the 146 amino acids of the beta chain of hemoglobin is replaced by lysine.
It is the predominant hemoglobin found in people with hemoglobin C disease.
The molecule structure is a2bC 2.
Hemoglobin C disease is relatively benign, producing a mild hemolytic anemia and splenomegaly.
Hemoglobin C trait is a benign disease.
Hemoglobin E:
This variant results from a single mutation in the hemoglobin beta chain.
This results from a single change of one amino acid, where glutamic acid at the 26th position of the 146 amino acids of the beta chain of hemoglobin is replaced by lysine.
Hemoglobin E is most prevalent in Southeast Asia (Thailand, Myanmar, Cambodia, Laos, Vietnam).
People with hemoglobin E disease have a mild hemolytic anemia and mild splenomegaly.
Hemoglobin E trait is a benign disease.
Hemoglobin Constant Spring:
Hemoglobin Constant Spring is a variant in which a mutation in the alpha globin gene produces an alpha globin chain that is abnormally long.
It is an abnormal hemoglobin having an extended polypeptide chain (31 additional amino acyl residues) on the α chain (thus, the α chain is 172 amino acids long instead of 141).
Approximately 20 % of the patients with Hb H disease also have this defect.
The designation hemoglobin Constant Spring derives its name from the isolation of the hemoglobin variant in a family of ethnic Chinese background from the Constant Spring district of Jamaica.
Hemoglobin H:
Hemoglobin H is a beta-4 tetramer which occurs when there are excess beta chains produced in the presence of defective alpha chain synthesis.
In hemoglobin H, only one gene for the production of alpha chain production has been inherited.
Hemoglobin Barts (Hb Barts):
Hemoglobin Barts develops in fetuses with four-gene deletion alpha thalassemia.
Hemoglobin Barts consists of four gamma chains.
Most individuals with four-gene deletion thalassemia and consequent hemoglobin Barts die in utero (hydrops fetalis).
The abnormal hemoglobin seen during fetal development in individuals with four-gene deletion alpha thalassemia was characterized at St. Bartholomew’s Hospital in London. The hospital has the fond sobriquet, St. Barts, and the hemoglobin was named “hemoglobin Barts.”
Although hemoglobinopathies and thalassemias are two genetically distinct disease groups, the clinical manifestations of both include anemia of variable severity and variable pathophysiology.
The hemoglobinopathies, or structural Hb variants, are attributable to amino acid substitution in either α or non-α chain.
More than 700 hemoglobinopathies have been described to date, the majority of which are clinically benign.
The clinically significant hemoglobinopathies are attributable to amino acid substitutions, primarily in the non-α chain.
Abnormal Hemoglobins
Hemoglobin S: a single change of one amino acid, where glutamic acid at the sixth position of the 146 amino acids of the beta chain of hemoglobin is replaced by valine (a2bS 2).
Hemoglobin C: a single change of one amino acid, where glutamic acid at the sixth position of the 146 amino acids of the beta chain of hemoglobin is replaced by lysine (a2bC 2).
Hemoglobin E: a single change of one amino acid, where glutamic acid at the 26th position of the 146 amino acids of the beta chain of hemoglobin is replaced by lysine (a2bE 2).
Hemoglobin Constant Spring: an abnormal hemoglobin having an extended polypeptide chain (31 additional amino acyl residues) on the α chain (thus, the α chain is 172 amino acids long instead of 141).
Hemoglobin H: a beta-4 tetramer which occurs when there are excess beta chains produced in the presence of defective alpha chain synthesis. It is a form of alpha thalassemia with only one (out of four) alpha chain genes being present.
Hemoglobin Barts (Hb Barts): hemoglobin Barts develops in fetuses with four-gene deletion alpha thalassemia. It consists of four gamma chains.
Compound heterozygous conditions:
These results from inheriting two different variant genes from the alpha globin gene cluster or two different variant genes from the beta globin gene cluster.
The nature of two genes inherited determines whether a clinically significant disease state develops.
Abnormal Hemoglobins
Hb D-Punjab: an abnormal Hb with a single β chain substitution, glutamine for glutamic acid (Glu >Gln) in the beta globin chain (molecular formula α2Aβ2121Glu → Gln).
Hb O-Arab: hemoglobin O Arab is a beta globin chain mutation (α2β2 121 Glu → Lys).
Hb G-Philadelphia: this is a common alpha globin chain variant [α68(E17) Asn >Lys].
Hb Hasharon: Hb Hasharon is a hemoglobin variant with an alpha globin mutation. Hemoglobin Hasharon (α2 47 Asp → His β2).
Hb Korle-Bu: the amino acid aspartic acid at the 73rd position of the beta chain is replaced by asparagine.
Hb Lepore: the Hb Lepore variant, consisting of two normal alpha globin chains (HBA) and two delta beta globin fusion chains which occur due to a “crossover” between the delta (HBD) and beta globin (HBB) gene loci during meiosis.
Hb M: a group of abnormal hemoglobins in which amino acid substitutions take place in either the alpha or beta chains but near the heme iron. This results in facilitated oxidation of the hemoglobin to yield excess methemoglobin. Specific types include Hb MIwate, α87His→Tyr (α chain, position 87, histidine replaced by tyrosine); Hb M Hyde Park, β92His→Tyr; Hb MBoston, α58His→Tyr; Hb MSaskatoon, β63His→Tyr; and Hb MMilwaukee-1, β67Val→Glu.
Hb Kansas: in Hb Kansas, threonine replaces asparagine in position 102 of the β chain.
3.4 Sickle Syndromes
The sickle syndromes are a group of hemoglobinopathies with the common feature of having at least one gene that produces hemoglobin S.
Sickle cell disease (SCD) and its variants are genetic disorders resulting from the presence of a mutated form of hemoglobin, hemoglobin S (HbS).
SCD denotes all genotypes containing at least one sickle gene, in which HbS makes up at least half the hemoglobin present.
In this book, the term sickle cell disease is used to denote all hemoglobinopathies having at least one gene that produces hemoglobin S.
The term sickle cell disease should not be used to denote sickle cell anemia.
Sickle cell anemia denotes homozygous sickle cell (HbSS).
There are many different types of sickle cell disease, but hemoglobin SS, hemoglobin SC, and sickle beta-thalassemia are the most common types of sickle cell disease.Related posts:
 Hydroxyurea Treatment for Sickle Cell Anemia
Hematopoietic Stem Cell Transplantation for Patients with Sickle Cell Anemia
Hydroxyurea Treatment for Sickle Cell Anemia
Hematopoietic Stem Cell Transplantation for Patients with Sickle Cell Anemia
 Cardiovascular Complications of Sickle Cell Anemia
Cardiovascular Complications of Sickle Cell Anemia
 Acute Appendicitis and Sickle Cell Anemia
Acute Appendicitis and Sickle Cell Anemia
 Genetics and Pathophysiology of Sickle Cell Anemia
Genetics and Pathophysiology of Sickle Cell Anemia
 Hepatobiliary Complications of Sickle Cell Anemia
Hepatobiliary Complications of Sickle Cell Anemia

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


