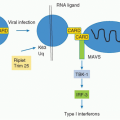
evolution.13 Conservation of gene structure of the TNF ligands outside these MHC paralogs is limited. An evolutionarily conserved pathway in Drosophila melanogaster, Eiger-Wengen system, is structurally and functionally related to the TNFSF-related signaling pathway, although this system is predominantly expressed in the nervous system in invertebrates.14,15,16
TABLE 27.1 Tumor Necrosis Factor Superfamily Chromosomal Locations | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 27.2 Tumor Necrosis Factor Receptor Superfamily | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
that residues in CRD2 and 3 of TNFR1 contact the ligand. Variation in binding interactions has been identified; for example, the receptors for BAFF have one functional CRD.2,19
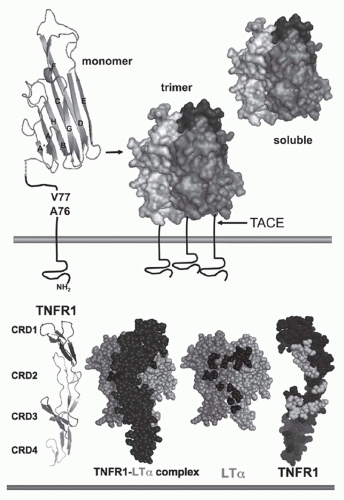 FIG. 27.1. Structure of Tumor Necrosis Factor (TNF) and TNF Receptors (TNFRs). Top: TNF. The β-sandwich of the TNF monomer (1A8M.pdf) (shown as a ribbon diagram) contains two stacked β-pleated sheets each formed by five antiparallel β strands (wide ribbons given letter designations, A-G) that fold into a Greek key or “jelly-roll” topology.219 The inner β-sheet (strands A, A′, H, C, and F) is involved in contacts between adjacent subunits, which promotes assembly into a trimer. The trimer is formed such that one edge of each subunit is packed against the inner sheet of its neighbor. The outer β sheet (strands B, B′, D, E, and G) is surface exposed. The trimeric structure is characteristic of all TNF-related ligands. The type II configuration of TNF (N-terminus inside the cell) anchors TNF to the membrane. The TNF trimer is ˜60 Å in height with a relatively flat base residing close to the membrane, resembling a bell-shape (shown as surface representation with different shades of gray used for each subunit of the trimer). The surface exposed loops between A-A′ and D-E strands are involved in receptor binding. TNF is cleaved by TNFα-converting enzyme (TACE), a member of the ADAM family of metalloproteinases (ADAM17) involved in processing of many cell surface proteins. TACE is a type 1 transmembrane protein that cleaves TNF between residues Val77 and Ala76, when all three sites in the trimer are cleaved TNF is released from the membrane. Bottom: TNFR and ligand complex. The ectodomain of TNFR1 forms an elongated molecule with CRD1 proximal to the N-terminus (ribbon diagram). The face of TNFR1 on the left engages LTα. In the ligand-receptor complex, the elongated receptor (dark) lies along the cleft formed between adjacent subunits. Shown (space-filling) is a single TNFR1 in complex with two subunits of LTα (lighter shades); the receptor N-terminus points upward, closest to the base of the ligand (transorientation). In the exploded view, TNFR1 is removed from in front of the ligand revealing the contact residues in the ligand, which are primarily located in the D-E and the A-A″ β-strands (dark shade). TNFR1 is rotated 180 degrees, exposing the contact residues in the receptor (light shade). Structures from 1TRN.pdf17 as visualized with MacPyMOL (www.pymol.org). |
feature bestowed in part by the propensity of the cytosolic tails to self-associate.25 In the physiologic setting, the expression level and compartmentalization of these receptors are tightly controlled. A subregion in the first CRD of TNFR1 known as the preligand assembly domain may restrict the orientation of the unligated receptor to prevent spontaneous activation.26 It is not known if this mechanism applies to all TNFRSF. Interestingly, some patients with periodic fever syndrome have mutant TNFR1 that form abnormal disulfide-linked oligomers that are retained intracellullarly and provoke misfolded protein response.27
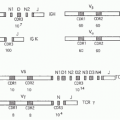
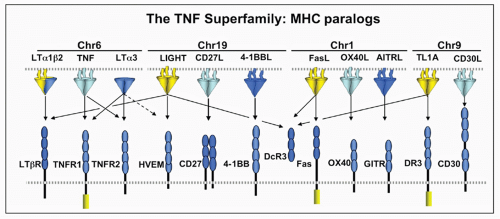 FIG. 27.2. The Tumor Necrosis Factor (TNF) Superfamily-Major Histocompatibility Complex (MHC) Paralogs. Members of the TNF ligand superfamily (above) and their corresponding receptors (below) are identified by connecting arrows. The ligands are grouped according to their chromosomal locations in the MHC paralogous regions. The number of cysteine-rich domains are depicted for each TNF receptor, and TNF receptors containing a death domain are identified by cylinder in the cytoplasmic tail. (Modified from Ware336). |
The engagement of ligands distinct from TNFSF members implicates a higher level of integration with other signaling pathways.
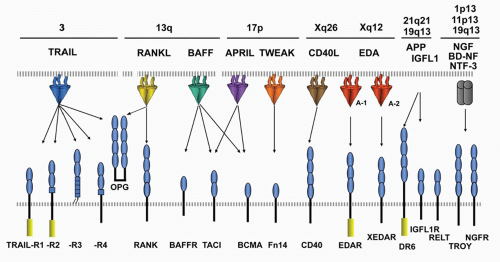 FIG. 27.3. The Tumor Necrosis Factor (TNF) Superfamily—Part 2. Members of the TNF ligand superfamily (above) and their corresponding receptors (below) are identified by connecting arrows. The ligands are grouped according to their chromosomal locations. The number of cysteine-rich domains are depicted for each TNF receptor, and TNF receptors containing a death domain are identified by cylinder in the cytoplasmic tail. (Modified from Ware336). |
TABLE 27.3 Viral Orthologs and Modulators of the Tumor Necrosis Factor Superfamily | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
for TNF is short, controlled by an adenylate-uridylate-rich element in the 3′ untranslated region.47 Deletion of the adenylate-uridylate-rich element in TNF mRNA in mice leads to a profound inflammatory disease.47,48 TNF mRNA is inducible in macrophages by multiple pathways, particularly innate activation pathways such as toll-like receptor signaling, whereas other ligands like 41BBL and OX40L are constitutively expressed in differentiated antigen-presenting dendritic cells (DCs). T- and B-lymphocytes require activation prior to expression of TNF. In these cells, signals via the antigen receptor utilizing nuclear factor of activated T cells (NFAT) transcription factors are needed for the induction of TNF transcription. The inducible or constitutive patterns of expression are also observed with some receptors as well.
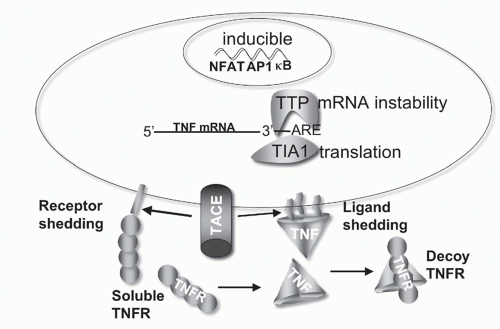 FIG. 27.4. Regulation of Tumor Necrosis Factor (TNF) Bioavailability. The expression of TNF is regulated at the transcriptional and translational levels, and bioavailability by altering its physical location and cellular receptors. TNF transcription is regulated by the action of multiple transcription factors including nuclear factors of activated T cells (NFAT), activated protein-1 (AP1), and NF-κB. NFAT is a predominant acting transcription factor regulating TNF transcription in T- and B-lymphocytes, and NF-κB and AP1 are important in myeloid lineage cells following activation via innate pattern recognition receptors, such as the toll-like receptors. TNF messenger ribonucleic acid (mRNA) stability is controlled by an adenylate-uridylate-rich element (ARE) in the 3′ untranslated region present in many transiently expressed inflammatory genes.337 Stability of TNF mRNA is decreased by the action of tristetraprolin, and T-cell intracellular antigen silences translation of TNF mRNA through the ARE.338 TACE proteolytically controls TNF at the membrane, generating the soluble form of TNF. TACE also cleaves TNFR1 and 2,339 downregulating cell surface receptors and releasing soluble receptors that retain TNF binding activity. Soluble TNFR can stabilize the TNF trimer at sub saturating concentrations, and at higher, saturating concentrations act as decoys competing for TNF binding to cellular receptors.340 |
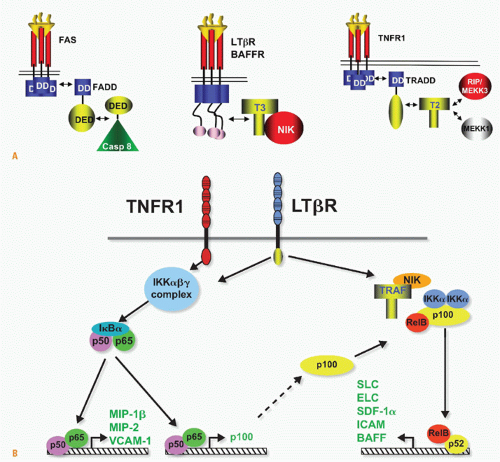 FIG. 27.5. Signaling Pathways and NF-κB. A: Adaptors link tumor necrosis factor receptors (TNFRs) to proteinases and kinases (upper panel). Three basic schemes link activated TNFRs to signaling pathways: the death inducing signaling complex formed with Fas is initiated by ligand clustering of Fas, promoting death domain (DD) interactions, which recruits Fas-associated death domain (FADD) (heterotypic interaction). The death effector domain of FADD links to the death effector domain of procaspase 8. The proximity of multiple procaspase 8 domains forms an active enzyme that can process other caspase 8 molecules. By contrast, TNFRs bind TNF-associated factor (TRAF) adaptors via short peptide motifs that release TRAF from associated kinases, such as NF-κB-inducing kinase (NIK). The third scheme is a combination of DD and TRAF recruitment. TNFR1 uses the DD protein TRADD to recruit RIP and TRAF2, promoting the activation of NF-κB. There are currently seven TRAF members with distinct interaction patterns with the TNFR family. TRAF proteins may function as regulators of key kinases. TRAF2, 3, and 6 function as modulators of several different kinases involved in toll-like receptor signaling, induction of type 1 interferon responses, and signaling by some TNFR family members. TRAF proteins contain an N-terminal RING finger motif, a coiled coil domain (isoleucine zipper), and the receptor association domain (TRAF) domain. The TRAF are trimers formed through their TRAF and coiled domains. The TRAF domain can bind to several different TNFR through a relatively short proline-anchored sequence that is responsible for binding directly to the mushroom head of various TRAF molecules.341 The zinc RING of TRAF6 functions together with Ubc13 and Uev1A as ubiquitin E3 ligase targeting proteins for proteosome degradation. TRAF2, 3 and cIAP form the ubiquitin E3 ligase that targets NIK. Modified with permission from Ware CF. Tumor necrosis factors. In: Bertino JR, ed. Encyclopedia of Cancer. San Diego, CA: Academic Press, Inc.: 2002;475-489. B: NF-κB activation. TNFR1 and LTβR induce distinct forms of the NF-κB family of transcription factors. TNFR1 signaling is a potent activator of RelA/p50 but does not activate the RelB pathway, whereas lymphotoxin βR can activate both. The RelA and RelB forms of the κB family control transcription of distinct sets of genes; however, the two pathways are interrelated through the control of p100 expression by the RelA/p50 complex. Modified from Dejardin et al.66 |
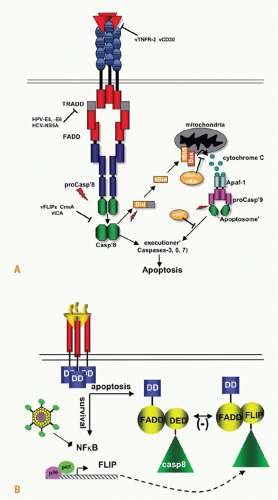 FIG. 27.6. Apoptosis and Survival Signaling. A: Ligation of tumor necrosis factor (TNF) receptor 1, Fas, or TNF-related apoptosis inducing ligand 1 and 2 activates apoptosis pathways. The nascent death-inducing signaling complex can directly cleave the executioner caspases 3, 6, and 7. Caspase 8 also cleaves BID to tBID, activating the mitochondrial regulated apoptotic pathway, dramatically accelerating cellular death. A number of viral proteins interfere with apoptosis induced through TNF pathway including human papilloma virus, hepatitis C virus, herpesvirus vFLIP, and virus orthologs of BCL2 and vMIA act on the mitochrondria. Adapted with permission.111 B: The components of the apoptotic pathway are preformed in the cytosol enabling the cell to respond rapidly to death signaling. In contrast, the key regulators of the apoptotic pathway, including Bcl2 and inhibitor of apoptosis, require new gene expression controlled by NF-κB. An important NF-κB-induced gene is cellular FLIP, which contains a pseudocaspase domain. The cFLIP protein interferes with caspase 8 activation, blocking the prodeath pathway. If the cell is transcriptionally inactive or unable to activate NF-κB, which often occurs in a pathogen-infected cell, then the proapoptotic pathway dominates the signals emanating from the TNF receptor 1 signaling complex leading to apoptosis and curtailing parasitism of the cell. |
coordinate the physiologic responses during inflammation (Table 27.4). The responses will reflect the characteristic of the inflamed organ, the local or systemic source of TNF, and the duration of TNF signaling. Acute inflammatory responses involve rapid changes in hemodynamics (plasma leakage and edema) and leukocyte adherence, extravasation, and organ infiltration induced by TNF. TNF production during chronic inflammation may contribute to systemic metabolic derangements and wasting (cachexia) or loss of organ structure and function (bone erosion in joints of patients with rheumatoid arthritis [RA]). In some models, TNF can promote an inflammatory environment that promotes tumor formation.73,74,75
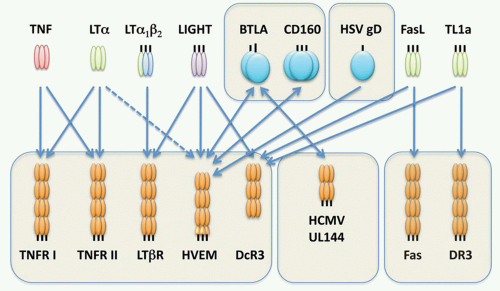 FIG. 27.7. The Immediate Tumor Necrosis Factor (TNF) Lymphotoxin (LT) Network. The cartoon depicts the signaling network formed between TNF, LTα, LTβ, and LIGHT, and their receptors. Each arrow indicates a ligand-receptor interaction. The network is defined by the extensive cross-utilization of ligand and receptors. Herpesvirus entry mediator (HVEM) forms a switch between positive cosignaling through LIGHT-HVEM interaction and inhibitory signaling through B- and T-lymphocyte attenuator (BTLA). LIGHT bound to HVEM activates TNF receptor-associated factor-dependent activation of NF-κB, whereas HVEM-BTLA acts through an immunoreceptor tyrosine-based inhibitory motif of BTLA to recruit the phosphatase SHP2, attenuating kinases activated by T-cell receptor signaling. The herpes simplex viron envelope protein gD attaches to HVEM acting as an entry step for infection. UL144 gene of human cytomegalovirus binds BTLA, but not LIGHT, selectively mimicking the inhibitory pathway of HVEM-BTLA. Fas Ligand and TL1A are included in this network through their interaction with decoy receptor-3. |
activation pathway, including TRAF6, NIK, IKKα, and Rel B, and their gene targets. Target genes including CXCR5, the receptor for CXCL13, show defective lymphoid organ structure, as do CCR7-/- mice (receptor for CCL19/CCL21). The developmental program of secondary lymphoid organ formation initiates at 9 days post coitus progressing in organized fashion from dorsal to lateral movement, with intestinal Peyer’s patches forming during the first postnatal week.83 The defect is irreversible in that transferring LTαβ-sufficient bone marrow into an adult LT-mutant mouse failed to induce lymph node formation. Accumulating studies indicate that the formation of lymphoid organs involves two distinct cell types, an embryonic LTβR-expressing mesenchymal stromal cell that responds to LTαβ expressed in a cell of hematopoietically derived lineage, termed the lymphoid tissue inducer cell. The lymphoid tissue inducer cell develops separately from lymphocytes and myeloid cells in a pathway dependent on ID2 and RORγt and different cytokines including interleukin (IL)7, RANK ligand (TRANCE, TNFSF11), and TNF. These cytokines induce surface LTαβ in lymphoid tissue inducer cells that differentially engenders formation of lymph nodes and Peyer’s patches.84 Cells of the lymphoid tissue inducer lineage are maintained in the adult tissues at low levels (0.5%), presumably aiding in the homeostasis of lymphoid organs.85,86
TABLE 27.4 Physiologic Correlates of Tumor Necrosis Factor-Mediated Gene Induction | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
TABLE 27.5 Phenotypes in Mice Deficient in Lymphotoxin and Tumor Necrosis Factor Immediate Family | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
rejection, or microbial infection often contain organized accumulations of lymphocytes reminiscent of secondary lymphoid organs, called tertiary lymphoid organs. Activated T and B cells provide the source of LTαβ that helps drive the process of forming these structures, but as antigen is cleared, immune responsiveness subsides and these structures resolve. A gradation of features may be found in the tertiary lymphoid organs including presence of DCs, expression of chemokines, high endothelial venules, segregated regions of T and B cells, and germinal centers, but these structures typically lack the permanence of a lymph node. TNF also contributes to formation of granuloma that assists in walling off bacteria.96 Thus, the LTαβ and TNF pathways operative in embryonic life also play critical roles in the adult in the formation of tertiary lymphoid structures.
receptors such as Fas and TNFR1 are thought to be involved in clonal contraction through apoptosis of activated effector cells, although TNF via TNFR2 also shows costimulatory action in naïve T cells. In tissue culture models, several of the TNF-related signaling pathways show costimulatory activities for T cells, which probably reflects the common induction of NF-κB-dependent survival genes, a common trait of TNFRSF members. However, analyses of physiologic models using genetically deficient mice reveal distinct roles for these molecules in the life cycle of T cells. Cosignaling systems are emerging as important targets to enhance immune responses to tumors or attenuate autoimmune diseases.114,125,126,127,128,129
TABLE 27.6 Lymphotoxins in Host Defense: Mouse Models
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
|---|