Fig. 11.1
Krebs cycle gene mutations in pheochromocytoma and paraganglioma and their downstream cellular effects. Mutations in succinate dehydrogenase (SDH) subunit and assembly factor genes, fumarate hydratase (FH), and mitochondrial malate dehydrogenase (MDH2) have been shown to predispose to chromaffin cell tumors. Due to their disruptive effect on central carbon metabolism, intracellular metabolites are altered leading to profound changes in downstream signaling pathways. Succinate and fumarate are known to inhibit α-ketoglutarate-dependent dioxygenases, including prolyl hydroxylases and histone and DNA demethylases. Succinate dehydrogenase also participates in the respiratory chain, where complex disruption can increase reactive oxygen species (ROS) production
Succinate dehydrogenase not only participates in the metabolism of carbon but is also a component of the electron transport chain located in the mitochondrial membrane (complex II) that is required for ATP generation through oxidative phosphorylation. Inhibition of complex II is known to induce oxidative stress in cell culture models [22, 28]. ROS, dependent on their equilibrium to antioxidants, can cause damage to intracellular molecules, including nucleic acids, proteins, and lipids [49]. DNA oxidation generates 8-hydroxy-2′-deoxyguanosine, a compound capable of inducing mutations and essentially leading to genomic instability. Oxidation of proteins may lead to functional impairment due to structural changes. ROS have been shown to promote different aspects of tumor development and progression, including proliferation, invasion and metastasis, and angiogenesis.
11.4 Tumor Microenvironment and Metabolism
An important component of solid tumors is the stromal microenvironment consisting of several cell types, such as fibroblasts, endothelial cells, and immune cells. Immunological staining studies of colorectal tumor tissue section for different metabolic enzymes (lactate dehydrogenase, pyruvate dehydrogenase, pyruvate-dependent kinase) and lactate and glucose transporters have indicated that fibroblasts and untransformed colorectal cells favor aerobic metabolism of pyruvate, whereas tumor cells have a suppressed aerobic metabolism aiding in survival under low oxygen conditions [31]. Based on these findings, the authors proposed a model of complementary metabolic pathways between highly glycolytic cancer cells, tumor-associated fibroblasts with strong lactate uptake and oxidation fueling aerobic metabolism, and tumor-associated endothelial cells with a high capacity for glucose uptake and aerobic pyruvate metabolism, but resistance to lactate intrusion. Other studies have demonstrated a metabolic symbiosis of tumor cells along oxygen and nutrient gradients with hypoxic cells being highly glycolytic and excreting lactate, which is then taken up as a carbon source by oxygenated cells [48]. Such cell-cell lactate shuttles have also been described in normal physiological tissues, including between neurons and astrocytes in the brain [15].
In further support of the aforementioned tumor microenvironment intercellular shuttling mechanisms, it has also been shown that SDHB silencing in a neuroblastoma model leads to increased proliferation, decreased glucose uptake, and increased lactate uptake compared to control [43]. Co-culturing these tumor cells with fibroblasts resulted in an additional increase in proliferation and lactate uptake, but also in a further decrease in glucose uptake.
11.5 Metabolic Biomarkers for Patient Stratification
Different underlying mutations in patients with PPGL result in tumors with distinct metabolic fingerprints. In PPGL, profiles of catecholamines and their metabolites, metanephrines, are characteristic for certain patient groups. Mutations affecting the Ras-Raf-MEK-ERK pathway, such as RET and NF1, lead to generally high tumor catecholamine contents and a relatively high proportion of epinephrine, whereas tumors due to VHL or SDHx aberrations predominantly produce norepinephrine with an additional high proportion of dopamine in those with SDHx mutations [16]. Patients with MAX mutations on the other hand present with an intermediate catecholamine phenotype [40]. SDHx-mutated tumors, which have a high risk of metastasis and recurrence, can be further distinguished by high levels of the dopamine metabolite 3-methoxytyramine [39]. Quantification of catecholamines and metanephrines by liquid chromatography tandem mass spectrometry (LC-MS/MS) therefore not only is crucial for the diagnosis of patients with PPGLs but also delivers information for stratification of patients [17].
Other biomarkers have been identified within the intermediates of the central carbon metabolism. Due to the blockade of the Krebs cycle at the site of SDH, PPGLs with disruptions in one of the SDH subunits have high levels of succinate and corresponding low levels of other Krebs cycle intermediates, including fumarate, citrate, isocitrate, and cis-aconitate [33, 45]. Measurements of the succinate-to-fumarate ratio by LC-MS/MS reliably identify tumors from patients with SDHx mutations making this a useful test for assessing the functional impact of mutational variants of unknown significance identified during genetic testing. In the same study, VHL-mutated tumors were also shown to have lower levels of fumarate, citrate, isocitrate, and cis-aconitate indicating increased requirements for biosynthesis leading to depletion of Krebs cycle intermediates. Citrate, a precursor for lipids, is known to be especially depleted in VHL-deficient RCC [21].
NMR-based studies identified a reduction of adenine nucleotides, glutamate, and N-acetylaspartate in SDHx-mutated PPGLs, as well as increased levels of glutathione and decreased levels of creatine in VHL-mutated PPGLs [27, 42]. Other metabolites altered in SDHx versus non-SDHx tumors include elevated methionine, glutamine, and taurine [26]. Recently, proton magnetic resonance spectroscopy was successfully employed to identify high succinate levels in patients’ tumors in vivo [54]. This technique can provide metabolic information in inoperable tumors, which could be used to predict tumor aggressiveness and guide therapeutic decision making and monitor therapeutic response.
11.6 Tumor Metabolism as a Therapeutic Target
Similar to cellular signaling cascades, the metabolism of tumor cells is distinct from that of healthy untransformed cells providing possibilities for targeted treatments. The Warburg phenotype of increased glucose uptake and utilization is one possible metabolic target. Gene silencing approaches and small molecule inhibitors were successfully used to inhibit different glycolytic steps in several cancer types leading to growth delay and apoptosis; however, due to lack of specificity, preclinical development of small molecule inhibitors is still ongoing [29]. Glutamine is another major carbon source used for oxidative phosphorylation and lipid biosynthesis or to replenish Krebs cycle intermediates. Glutamine dependence has been shown in different cancers, including VHL-mutated RCC and MYC-transformed tumors [21, 56]. Targeting glutamine uptake and metabolism through glutaminase is another proven concept for metabolic intervention.
A targeted treatment for SDHx-mutated tumor cells has been suggested by employing a genome-scale model of cancer metabolism [20]. This model predicted synthetic lethality for inhibition of pyruvate carboxylase (PC) in SDH- and FH-deficient cells. In the latter case, separate inhibition of nine other proteins was predicted to also selectively kill FH-mutated cells; these include seven enzymes involved in heme biosynthesis and two transporters. PC replenishes Krebs cycle intermediates by carboxylating pyruvate to oxaloacetate, which is essential for aspartate biosynthesis, a precursor for proteins, nucleotides, and other nonessential amino acids (Fig. 11.2). An alternative source of oxaloacetate is glutamine feeding into the Krebs cycle via α-ketoglutarate. The oxidative metabolic route is however blocked in SDHx-, FH-, and also MDH2-mutated tumor cells.
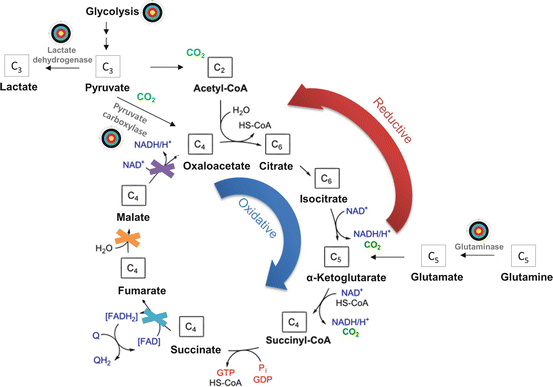
Fig. 11.2
Targeting tumor metabolism in pheochromocytoma and paraganglioma. There are several possible points of intervention in the central carbon metabolism (marked by bull’s eye). Glycolysis can be inhibited by restriction of glucose import via glucose transporters or by inhibition of specific enzymes, including lactate dehydrogenase. Tumors with mutations in succinate dehydrogenase, fumarate hydratase, or mitochondrial malate dehydrogenase (marked as crosses) are dependent on additional carbon sources to replenish Krebs cycle intermediates. Those could be pyruvate metabolized by pyruvate carboxylase or glutamine metabolized by glutaminase. Due to the blockade of oxidative α-ketoglutarate metabolism, tumors with the abovementioned mutations use reductive metabolism to replenish citrate and oxaloacetate pools. Hence, pyruvate carboxylase and glutaminase may be suitable targets for small molecule inhibitors
Cell culture models have since established that SDHB knockout cells are dependent on extracellular pyruvate to sustain maximal glycolytic flux. Pyruvate deprivation or PC knockdown results in a selective growth delay of SDHB knockout cells compared to wild type in vitro and as xenografts [7, 36]. PC protein levels were also found to be upregulated in SDHB-mutated compared to SDHB wild-type PPGLs. Experiments with 13C labels demonstrated that pyruvate carbons replenish oxaloacetate pools in SDHB knockout cells to maximize the glycolytic flux into the Krebs cycle via acetyl-CoA. This process supports the biosynthesis of aspartate from oxaloacetate. Glutamine is the major source of aspartate in SDHB wild-type cells, but only contributes to 20 % in SDHB-mutated cells. Compared to wild-type chromaffin cells, which preferably utilize oxidative glutamine metabolism, reductive carboxylation of glutamine is increased in SDHB knockouts indicating another potential point of intervention for targeted therapy.
Cardaci et al. furthermore showed that the respiratory chain in SDH-deficient kidney cells operates close to the bioenergetic limit and that these cells have a reduced coupling efficiency resulting in ATP being predominantly produced by glycolysis rather than oxidative phosphorylation. Inhibition of lactate dehydrogenase, a key glycolytic player, with oxamate selectively inhibited proliferation in SDHB-mutated cells by 40 %. By using 13C-labeled substrates, reduced entry of glucose carbons in the Krebs cycle was demonstrated leading to reduced citrate levels, which correlated with findings in PPGL tumor tissue.
Targeting the metabolism of tumors can be an effective way to selectively kill cancer cells by limiting their biosynthetic capacity. Clinical trials with metabolic inhibitors as single agents or in combination had varying successes, and many trials are still ongoing [46]. There is a strong need for the development of more specific agents targeting different metabolic pathways; a careful selection process to match certain drugs or drug combinations to specific patients will also be of importance to success of any therapies targeting tumor metabolism.
References
1.
Aspuria PJ, Lunt SY, Varemo L, Vergnes L, Gozo M, Beach JA, et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014;2:21.CrossRefPubMedPubMedCentral
2.
Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69(1):49–54.CrossRefPubMedPubMedCentral
3.
4.
5.
6.
Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19(15):3011–20.CrossRefPubMedPubMedCentral
Related posts:
 Molecular Genetics of Gastroenteropancreatic Neuroendocrine Tumours
Molecular Genetics of Gastroenteropancreatic Neuroendocrine Tumours
 Molecular Genetics of MEN1-Related Neuroendocrine Tumors
Molecular Genetics of MEN1-Related Neuroendocrine Tumors
 Radionuclide Imaging of Pancreatic Neuroendocrine Tumours
Radionuclide Imaging of Pancreatic Neuroendocrine Tumours
 Nanoparticles for Radionuclide Imaging and Therapy: Principles
Nanoparticles for Radionuclide Imaging and Therapy: Principles
 Radionuclide Imaging of Medullary Thyroid Carcinoma
Radionuclide Imaging of Medullary Thyroid Carcinoma
 Radionuclide Imaging of Gastrointestinal Neuroendocrine Tumors
Radionuclide Imaging of Gastrointestinal Neuroendocrine Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


