Tumor Immunology and Pediatric Cancer
Terry J. Fry
Paul M. Sondel
Crystal L. Mackall
The immune system is a complex network of cells and soluble factors that has evolved to respond to infection with foreign, pathogenic organisms. Its hallmarks are its ability to recognize danger, mobilize a multipronged effector response, and generate “immunologic memory” that renders future responses to the same organism more efficient. Cancer and the immune system interface at multiple levels. Immunodeficient patients have a higher incidence of cancer, and many cancer therapies are immunosuppressive, making infection a major comorbidity in cancer patients. Moreover, a dynamic microenvironment exists at the immune-tumor interface, which can permit incipient cancers to become immune evasive. Finally, immune-based therapies can provide clinically meaningful antitumor effects, with substantial progress seen over this past decade. After decades of promise, recent breakthrough discoveries have resulted in the designation of cancer immunotherapy as 2013’s “Breakthrough of the Year” by Science Magazine,1 making immunotherapy a centerpiece of the anticancer repertoire. Given the goal of finding less toxic, more effective therapies for childhood cancers, it is essential that the pediatric oncology community learn how to effectively apply current principles of tumor immunology to improve outcomes for children with cancer. This chapter will review the interface between cancer and the immune system, with emphasis on issues relevant to pediatric cancer.
OVERVIEW OF THE IMMUNE SYSTEM
The immune system can be divided into innate and adaptive immunity (Fig 5.1). Innate immune cells serve as the first line of defense, but do not express clonotypic receptors, and therefore are unable to mediate immune memory. The adaptive immune system, consisting of B cells and T cells, recognizes antigens with high specificity via clonotypic antigen-specific B-cell receptors (which are antibodies expressed on the B-cell surface) or T-cell receptors (TCRs), respectively. Many models have been developed to explain how the immune system can distinguish between foreign pathogens and self tissues. It is clear that during immune cell development (e.g., lymphopoiesis), the immune system becomes tolerant to self-antigens, through “clonal deletion” of T and B cells whose antigen-specific receptors recognize antigens expressed on normal autologous tissues (self-antigens). However, clinical and experimental data demonstrate that clonal deletion during lymphopoiesis is not complete, since healthy humans and mice harbor sizable numbers of T cells and B cells expressing receptors that recognize antigens expressed by self-antigens. Many antigens overexpressed on tumors, which are being used as targets for some forms of immunotherapy, are self-antigens that are expressed at high levels during development but present at lower levels on normal tissues in adult life. Thus the fact that clonal deletion is incomplete, leaving room for the induction of some “autoreactive” immune responses to some self-elements, including some tumor antigens, is directly relevant to tumor immunology and immunotherapy.
The presence of self-reactive lymphocytes is well illustrated by studies of immunotherapies administered to patients with melanoma. Most patients with malignant melanoma harbor melanoma-reactive T cells that recognize melanocyte differentiation antigens. These melanocyte differentiation antigens are highly expressed during melanocyte development and are expressed at low levels on normal melanocytes throughout life. The clonal deletion model predicts that cells reactive to such antigens would be completely eliminated during lymphopoiesis. However, a number of immunotherapeutic approaches, including administration of interleukin-2 (IL-2), tumor vaccines, or infusion of ex vivo-activated tumor-infiltrating lymphocytes (TIL), induce immune responses toward these melanocyte self-antigens, which often results in areas of melanocyte destruction, causing the clinical condition of vitiligo.
The “danger model” provides another construct to understand how immune tolerance is normally maintained despite the presence of self-reactive lymphocytes. This model hypothesizes that self-reactive lymphocytes are not activated to recognize (or destroy) self-tissue, unless they receive a second triggering signal, associated with some tissue damage (namely, a danger signal). This danger signal initiates the immune response by first activating innate immune responses, which are critical for stimulating adaptive T- or B-cell responses, thereby breaking self-tolerance. The innate immune system is composed of dendritic cells (DCs), neutrophils, monocytes, tissue macrophages, eosinophils, basophils, natural killer (NK) cells, and the complement cascade (Fig. 5.1). In addition to providing the first line of defense against infectious pathogens (see Chapter 40), innate immune cells play a central role in recognizing danger signals associated with infection and tissue inflammation, and then initiating, amplifying, and directing adaptive immune responses. Triggering of pattern recognition receptors, most notably toll-like receptors (TLRs) on innate immune cells by pathogen-associated molecular patterns (PAMPs) (such as bacterial cell wall components, lipopolysaccharide [LPS], viral RNA or DNA) or damage-associated molecular patterns (DAMPs) (released by dead or dying tissues), activates innate immune cells and initiates adaptive immune responses. DAMPs within neoplastic tissues may at times also induce innate immune responses and initiate adaptive immunity.
INNATE IMMUNE CELLS AND THE TUMOR MICROENVIRONMENT
Dendritic cells, macrophages, and immature myeloid cells recently described as myeloid-derived suppressor cells play significant roles in modulating antitumor immunity, both through local effects within the tumor microenvironment and through modulation of systemic immune responses in tumor-bearing hosts (Fig. 5.2). Although DCs are present in only trace amounts (1% to 2% of nucleated cells) in blood, bone marrow, and tissues, they are primary sentinels of the immune system. Their name derives from the fact that upon activation and differentiation, they develop dendritic-like projections that serve to increase the surface area of their cell membrane, which deliver essential signals to the adaptive immune system (Fig. 5.3). Substantial progress has been made in defining DC subsets and elucidating their ontogeny in humans, including the recent discovery of a separate dendritic cell hematopoietic progenitor lineage (reviewed in Ref 2).
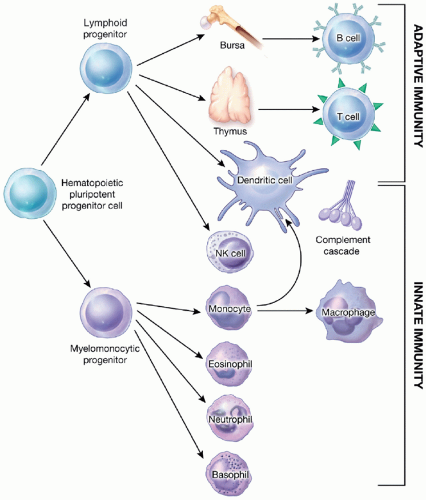 Figure 5.1 Innate and adaptive immune systems. All cells of the immune system are derived from pluripotent hematopoietic stem cells that give rise to two distinct progenitors: one for lymphoid cells and the other for myeloid cells. The common lymphoid progenitor has the potential to differentiate into T, B, NK, and lymphoid dendritic cells depending on its microenvironment (i.e., the thymus or bone marrow). Immune cells derived from primitive myeloid progenitors include phagocytic effectors (e.g., macrophages, neutrophils, and monocytes) and myeloid dendritic cells. It should be emphasized that the distinction between these systems is somewhat artificial since there is significant cross talk between innate and adaptive immune effectors during the initiation and completion of the immune response. |
DCs are defined by expression of class II major histocompatibility complex (MHC) molecules on a cell that does not express lineage markers associated with T, B, NK, monocyte, or RBC differentiation. The two major circulating DC subsets are plasmacytoid DCs (pDCs) and classical DCs (cDCs). In tissues other subpopulations of DCs are also found, including Langerhans cells (dendritic cells that populate the epidermis), CD14+ DCs, CD1a+ DCs, and thymic DCs. DCs reside in essentially every tissue of the body, where they continually survey the antigenic milieu. In the absence of inflammation, DCs do not initiate immune responses but rather contribute to immune tolerance to self-antigens through as yet poorly understood mechanisms. However, upon contact with inflammatory signals, immature DCs differentiate into mature DCs, characterized by high-level expression of MHC class I, class II, costimulatory molecules, as well as IL-12 production that drive T-cell activation, and direct T-cell responses. T cell and DC cross talk also follows, with CD40L on activated CD4+ cells upregulating B7-1, B7-2, and MHC molecules on DCs.
Both plasmacytoid DCs (pDCs) and myeloid DCs (mDCs) can be generated in vitro by incubating monocytes with granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 or by incubating pluripotent hematopoietic progenitor cells with GM-CSF and/or flt3 ligand and can be matured using a variety of agents. This has greatly aided the study of these normally rare cells and has also provided opportunity for clinical applications of DC-based immunotherapy for cancer. Dendritic cells are essential for immunologic priming of the host to tumor-associated antigens. This requires that DCs infiltrate the tumor, take up and process tumor antigens into peptides, then travel to the tumor-draining lymph node, where they present peptides in the context of costimulation to T cells. Consistent with this model, improved outcome is associated with higher levels of dendritic cell infiltration in some cancers.3,4,5,6
Macrophages and immature myeloid cells also infiltrate tumors and generally serve to suppress antitumor immune responses. Macrophages are short-lived, monocyte-derived, tissue-resident cells that phagocytize cells and debris, but are poor antigen presenters and do not express adequate levels of class II MHC or costimulatory molecules to initiate T-cell responses. Depending upon the immune milieu, macrophages are either “classically activated” (M1) or “alternatively activated” (M2), which parallels Th1 and Th2 T-cell differentiation.7 Acute inflammation involving TLRs and IFNγ induce M1 activation; this may result in macrophages capable of mediating antitumor effects. However, the vast majority of tumor-associated macrophages (called TAMs) undergo alternative activation, are immunosuppressive, and facilitate tumor growth by augmenting angiogenesis. As a result, macrophage infiltration in tumors correlates with diminished survival.8,9 Compared to adult cancers, pediatric tumors are predominantly infiltrated by macrophages, whereas dendritic cell and lymphocyte infiltration represent a minor component of the inflammatory milieu.10 This histologic observation suggests that inherent immune recognition of tumors, followed by their immune destruction (a process designated immune surveillance), may be less efficient in the setting of pediatric cancer than many more common cancers of adults.
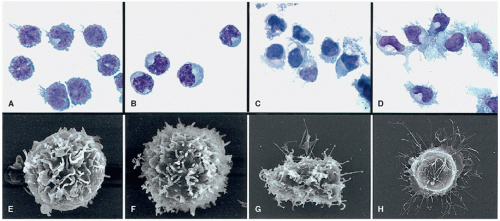 Figure 5.2 Morphology of DC subsets. Rhesus pre-DC subsets were stained with Giemsa (A-D) or examined using scanning electron microscopy (E-H) before and after culture with DC maturation-inducing factors GM-CSF and CD40L. A: Freshly sorted CD11c+ premyeloid DCs showing a high nucleus-cytoplasm ratio, reniform or multilobulated nuclei, and few prominent dendrites. B: Freshly sorted CD123++ prelymphoid DCs with a typical prominent Golgi region and lateralized reniform nucleus. C: After culture with rhGM-CSF and rhCD40L for 24 hours, CD11c+ premyeloid DCs showed abundant and well-developed dendrites. D: After culture with rhIL-3 and CD40L for 3 days, CD123++ pre-DCs also acquired striking dendritic morphology. (E-H) SEM images of cells corresponding to populations in panels A to D. Original magnifications: ×400 (A-D), ×7500 (E), ×9000 (F), ×5000 (G), and ×3500 (H). (From Coates PT, Barratt-Boyes SM, Zhang L, et al. Dendritic cell subsets in blood and lymphoid tissue of rhesus monkeys and their mobilization with flt3 ligand. Blood 2003;102:2513.) |
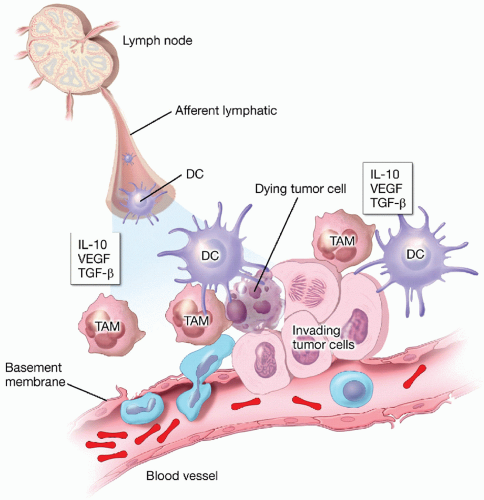 Figure 5.3 The immune-tumor interface. The microenvironment of a growing tumor represents a complex and dynamic interaction between an invading tumor and the inflammatory/immune response. Tumor invasion through the basement membrane is required for metastasis and predicted to induce activation of innate immune effectors that transit from the blood to tissue. If local inflammation is sufficient to activate resident dendritic cells, tumor antigens will be acquired and transported to draining lymph nodes, where T-cell activation can occur. If immunosuppressive mediators predominate, local inflammation is prevented and immune priming does not occur. Immunoactivating elements including heat shock proteins (HSPs; elaborated from dying tumors) and DC-activating receptors present on the surface of apoptotic and necrotic tumor cells. Immunosuppressive elements include soluble mediators released by the tumor, including IL-10, VEGF, and TGF-β. TAMs are tumor-associated macrophages that contribute to local immunosuppression. |
Myeloid-derived suppressor cells (MDSCs) are immature myeloid cells (both monocytic and granulocytic) that expand in tumor-bearing hosts in response to a wide array of tumor-derived factors. They induce immune suppression largely via production of inducible nitric oxide synthase and arginase,11,12 agents that can have direct suppressive effects on the effector functions of immune cells and give rise to immunosuppressive TAMs and tumor-associated neutrophils. Recent studies have documented the expansion of immunosuppressive myeloid-derived suppressor cells in patients with pediatric cancer,13 and animal models demonstrate that interfering with trafficking of these cells to the tumor can augment the effectiveness of immune-based therapies.14 It is likely that standard chemotherapy regimens administered to children with cancer have important effects on MDSCs, both transient depletion associated with myelosuppression and potentially increased levels of these immunosuppressive cells associated with endogenous or pharmacologically administered granulocytic growth factors. Several soluble mediators (IL-10, vascular endothelial growth factor, and transforming growth factor-β) also contribute to an immunosuppressive tumor microenvironment. In summary, the activation of innate immune responses is essential if adaptive immunity is to be induced within the tumor microenvironment, and immunosuppressive innate immune cells play important roles in suppressing antitumor immune responses.
T LYMPHOCYTES AND CO-STIMULATION
T lymphocytes (designated “T” for their thymic origin) evolved primarily to eradicate viral infections; however, T cells also play central roles in autoimmunity, rejection of transplanted tissues, and antitumor immune responses. The TCR complex is organized into two major elements, one for antigen-specific recognition (the αβ or γδ TCR) and the other for cell activation (CD3) following antigen recognition (Fig. 5.4). All mature T cells express identical CD3 molecules (containing γ, δ, ε, and ζ chains). For approximately 95% of T cells, the antigen-specific TCR comprises a unique αβ heterodimer, whereas 5% of T cells recognize antigen through the γδ TCR. γδ T cells reside primarily in the gut, show limited TCR diversity, possess cytolytic capacity prior to activation, and can recognize virally infected cells in an MHC-nonrestricted manner, all features that are somewhat similar to the functions of NK cells. Like NK cells, γδ T cells likely serve as a first line of defense against viral and other pathogens.
The αβ TCR heterodimer endows T cells with fine specificity for antigen recognition, since peptides differing in only one amino acid often show widely disparate recognition by T cells. TCR genes are similar in structure to immunoglobulin genes. All nonimmune cells in any individual contain an identical pattern of αβ TCR genes maintained in the germline configuration, which is a linear arrangement of segments termed variable (V), diversity (for β chain only) (D), junctional (J), and constant (C) regions. During T-cell receptor gene rearrangement, the VDJ elements of both the α chain and the β chain of each unique αβ TCR first rearrange in different combinations; then “N segment additions” are added to further modify each rearranged chain. TCR rearrangement occurs during the early stages of thymocyte development and results in the TCR diversity, which is the hallmark of T-cell immunity. When TCR rearrangement is completed, each T cell expresses only one form of the rearranged αβ TCR, and all progeny of this T cell also express the identical rearranged αβ TCR.
From the enormous potential repertoire of TCR specificities that are randomly generated, a carefully controlled series of positive and negative selection events occur during thymopoiesis, resulting in the survival of a mere fraction of the T cells that begin the process. T cells are selected in the thymus largely on the basis of their TCR-binding affinity for self-MHC. TCR specificities showing intermediate affinity for self-MHC molecules are generally preferred, whereas those with very low or very high affinity do not survive. This maximizes the chance for efficient recognition of antigens of foreign pathogens (presented by self-MHC) while minimizing the chance for autoimmunity. Most T cells with high-affinity receptors for self-antigens are deleted. However, as discussed earlier, the process by which self-reactive cells are clonally deleted is not entirely complete, leaving a small component of circulating mature T cells that have retained some residual (but attenuated) capability to recognize antigens on normal autologous tissues (i.e., self-reactive T cells). Since many cancer antigens are overexpressed antigens found on certain normal cells (i.e., self-antigens), the repertoire of T cells available to recognize most tumor antigens (particularly those that are differentiation antigens) is relatively weak, compared with the repertoire of T cells that respond to truly foreign elements.
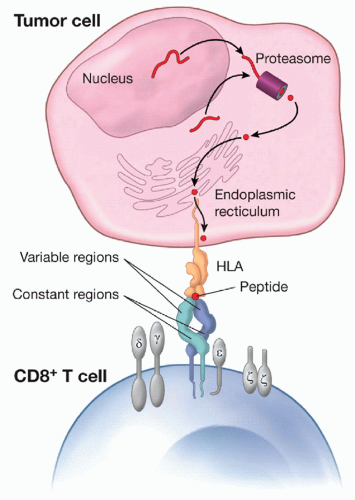 Figure 5.4 Peptide recognition by the TCR complex. The α- and β-polypeptide chains of the TCR are composed of variable and constant regions (shown in blue), which form a heterodimer anchored in the T-cell membrane. The heterodimer recognizes and binds to peptide associated with an HLA molecule on the surface of a presenting cell (shown in the figure as a tumor cell). The nonpolymorphic CD3 polypeptides (γ, δ, ε, and ζ) are assembled together with the αβ TCR and are involved in signal transduction. Peptides presented by HLA class I molecules are typically derived from the intracellular processing of endogenous proteins that are degraded in the proteasome and subsequently are transported to the endoplasmic reticulum, where they bind nascent MHC molecules prior to transport to the cell surface. |
Molecular analysis of rearranged TCR-DNA within T-cell populations is clinically useful for a variety of purposes. It can distinguish polyclonal reactive processes (i.e., a T-cell response to a viral antigen) from monoclonal malignant expansions (i.e., a T-cell malignancy), and molecular monitoring of the unique TCR rearrangements within T-cell malignancies using polymerase chain reaction or next-generation sequencing15 allows sensitive detection
of minimal residual neoplastic disease. Measures of the relative diversity within the TCR repertoire have also been used to evaluate the effectiveness of immune reconstitution as immune competence generally correlates with greater TCR diversity.
of minimal residual neoplastic disease. Measures of the relative diversity within the TCR repertoire have also been used to evaluate the effectiveness of immune reconstitution as immune competence generally correlates with greater TCR diversity.
The αβ TCR recognizes peptides derived from cellular proteins, which become accessible on the surface of the target cell via binding to MHC molecules. Human MHC molecules, called HLA (human leukocyte antigen), contain regions that are highly polymorphic (namely, regions that show many distinct amino acid sequence differences between different HLA alleles controlled by the same MHC locus) and regions that are nonpolymorphic (namely, regions that show very high levels of identity or homology between different alleles controlled by the same MHC locus). One nonpolymorphic region on virtually all class I HLA molecules (HLA-A, B, and C) binds to the CD8+ molecule (expressed primarily on cytotoxic T cells). A distinct nonpolymorphic region on MHC class II molecules (HLA-DR, DP, DQ) binds to the CD4+ molecule (expressed primarily on helper T cells). The polymorphic regions of the HLA molecules form a groove, which binds peptides with varying affinities. In order for a peptide to engage a reactive T cell, it must bind within the polymorphic groove of a particular HLA molecule (Fig. 5.4). Peptides bound to MHC class I are typically 9 to 11 amino acids in length, whereas MHC class II-binding peptides are typically 13 to 20 amino acids in length. Because binding of individual peptides varies greatly among the multitudes of MHC alleles that exist across individuals, individual TCRs recognize specific peptides only when presented by a correspondingly specific MHC molecule; they will not recognize the same peptide presented by a genetically distinct MHC molecule. In other words, when αβ-TCR-bearing T cells recognize any peptide, the peptide is specifically recognized only if it is presented in the groove of the corresponding MHC molecule that binds it; the TCR recognition of the peptide is restricted by the requisite presentation of that peptide by the MHC. This property has been termed MHC restriction and serves as a fundamental concept of T-cell immunology.
A detailed discussion of the biochemistry and the cell biology of antigen presentation by MHC molecules is beyond the scope of this chapter, but to summarize, endogenous cellular proteins undergo proteolysis and processing in the proteasome. Some peptides derived from this process of normal protein turnover are transported into the endoplasmic reticulum via chaperone molecules, called transporters of antigen processing, which transfer peptides to unstable MHC class I/β2-microglobulin dimers, thus giving rise to a stable complex that is transported and presented on the cell surface. Under normal circumstances, innumerable peptides derived from self-proteins are expressed continuously on all nucleated cells, since MHC class I is ubiquitous (Fig. 5.4). Despite this, in the absence of danger signals that activate innate immunity, immune responses are not induced, and self-tolerance is maintained. Unlike MHC class I molecules, which are expressed on all nucleated cells, MHC class II molecules are expressed only on specialized antigen-presenting cells (APCs), including DCs, monocyte-macrophages, B cells, activated T cells, and endothelium. MHC class II-expressing cells survey the environment for potentially dangerous or pathogenic stimuli; therefore, peptides presented by MHC class II include products derived from sampling the external milieu.
If TCR signaling (typically referred to as signal 1) is provided in the absence of activating costimulatory signals (signal 2), T cells become anergic and resistant to subsequent activation. Hence, signaling via the TCR is required for T-cell activation, but it is not sufficient. Signal 2 can be either stimulatory or inhibitory (Fig. 5.5), with signals augmenting T-cell activation described as costimulatory and signals preventing T-cell activation described as checkpoints. The most potent costimulatory signals for naive T cells are B7-1 (CD80) and B7-2 (CD86), which signal via CD28 on the surface of T cells, thereby enhancing proliferation and IL-2 production. In contrast, cytotoxic T-lymphocyte-associated protein 4 (CTLA4), a prototypic checkpoint molecule with high affinity for B7-1 and B7-2, competes with CD28 for binding with these molecules. CTLA4 activation diminishes T-cell activation, limiting the magnitude of the immune response and diminishing the likelihood of autoimmunity and lymphoproliferation. Congenital absence of CTLA4 in mice leads to lethal lymphoproliferation, and inhibition of CTLA4 during the course of an immune response amplifies immunity toward poorly immunogenic tumors in mouse models. Potent antitumor activity, with substantial clinical responses as well as clinically meaningful durable remissions have recently been seen in clinical trials for melanoma and other tumors in adults using checkpoint inhibitors, such as monoclonal antibodies (mAbs) that block CTLA4 binding to CD28. These are discussed later in the chapter, along with their implications for pediatric cancer treatment.
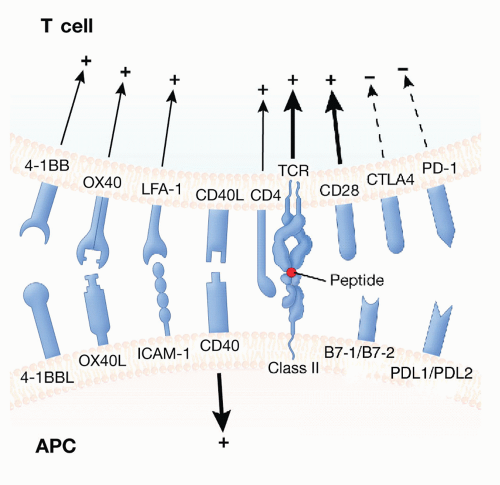 Figure 5.5 The T-cell immune synapse. Activation of T lymphocytes by APCs requires TCR signaling via peptides bound to HLA as well as costimulatory signals delivered by several potential receptor ligand interactions. Many positive costimulators that contribute to T-cell activation have been identified and are shown. A few negative costimulatory molecules (CTLA4, PD-1), also known as checkpoints, have also been described. CTLA4 acts as a negative regulator of T-cell activation and competes with CD28 for binding to CD80 and CD86 on APCs. CD40-CD40L interactions primarily signal the APC, which further upregulates costimulatory molecules. Additional interactions occur between CD4 molecule on the T cell and the nonpolymorphic region of the class II HLA molecule. 4-1BB is primarily a costimulator for CD8+ T cells, while OX40 is primarily a costimulator for CD4+ T cells. |
Once activated, T cells participate in immune responses via secretion of soluble mediators or through cell-to-cell interactions. CD4+ cells are described as helper T cells (Th) since they are rich sources of soluble mediators, including IL-2, a critical cytokine for amplifying CD8+ proliferation; IL-10, which dampens immunity by downregulating APC function; IL-4, which provides a critical signal for inducing humoral immunity; and IL-17, which may play an important role in antitumor immunity.16 A small subset of CD4+ T cells potently suppresses T-cell immune responses. These are known as regulatory T cells (Tregs). They are important in preventing autoimmunity. Treg cells constitutively coexpress CD25 (the α-chain of the IL-2 receptor [IL-2R]) and are distinct in function from activated Th, which transiently express CD25. Tregs suppress autoimmunity, graft-versus-host disease (GVHD),
and tumor immunity. Depletion of Tregs can augment antitumor immunity in several animal models. Remarkably, interleukin-2 (IL-2), which has been used as an immune stimulant for many years, selectively expands Tregs, and this immunosuppressive effect of IL-2 may serve to limit its effectiveness in the context of antitumor immunity.17
and tumor immunity. Depletion of Tregs can augment antitumor immunity in several animal models. Remarkably, interleukin-2 (IL-2), which has been used as an immune stimulant for many years, selectively expands Tregs, and this immunosuppressive effect of IL-2 may serve to limit its effectiveness in the context of antitumor immunity.17
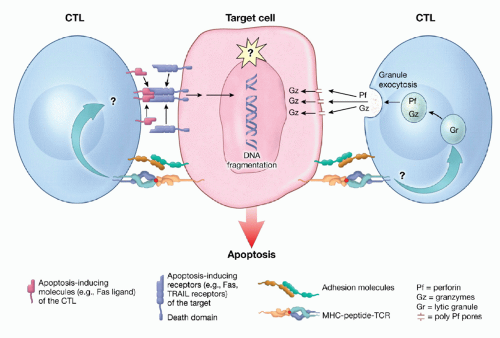 Figure 5.6 Mechanisms of lymphocyte-mediated cytolysis. Shown is a schematic diagram of the nonsecretory, receptor (e.g., Fas and TRAIL)-mediated (left) and the secretory, perforin/granzyme-mediated (light) mechanisms of lymphocytotoxicity. (Adapted from Berke G. Unlocking the secrets of CTL and NK cells. Immunol Today 1995;16:334.) |
CD8+ cells, termed cytolytic T lymphocytes (CTLs), serve primarily as effector populations, and have a potent capacity to kill target cells and to produce cytokines including IFN-γ. Activated cytolytic CD8+ cells can cause rapid cellular destruction through one of two pathways. Granule-mediated cytolysis involves perforin (a complement-like molecule) release, which first creates pores in the target membrane, followed by release of granzymes, which then efficiently enter the target cell and induce apoptosis. Alternatively, CD8+ cells can induce apoptosis through TNF receptor family members, including TNF, Fas, or TRAIL receptors (Fig. 5.6).18
B LYMPHOCYTES
The primary role of B lymphocytes (designated “B” for their bursal origin in fowl) is production of immunoglobulins, soluble molecules that specifically bind protein, glycoprotein, and carbohydrate antigens. Immunoglobulins bind to whole circulating proteins or molecules expressed on the surface of cells or pathogens. In contrast, T cells recognize peptides presented on the cell surface of MHC-bearing cells (discussed above) rather than intact proteins. Each immunoglobulin molecule consists of light (κ or λ) chains and heavy (µ, γ, α, δ, or ε) chains. Intact immunoglobulin molecules have a variable antigen-binding (Fab) end and a constant end (Fc) that can fix and activate complement and bind to Fc receptors expressed by a variety of cells. Memory B cells and plasma cells produce immunoglobulin for years after immunization; therefore, circulating antibody can provide rapid protection against second or subsequent exposure to antigenic pathogens.
Like TCR rearrangement that gives rise to exquisite T-cell diversity, developing B cells rearrange immunoglobulin genes to generate remarkable B-cell diversity. Immunoglobulin gene rearrangement involves selection from multiple segment possibilities within variable (V), diversity (D), and joining (J) regions. There are at least 40 VH regions; 27 D regions, with 3 different reading frames; and 6 JH regions, potentially resulting in 19,440 possible heavy chain combinations. Linking the V and J regions for each of the κ and λ light chains yields 265 different light chain combinations. Additional insertion of 1 to 6 nucleotides by TdT at each joining site adds to the myriad of possible combinations, estimated to be up to 1024 possible unique antibodies potentially generated from any one individual. Quantitative analyses suggest that the antibody repertoire number may be closer to 106 distinct unique B-cell clonotypes at any time.19 While a single B cell and its progeny can secrete many copies of a single specific immunoglobulin molecule, different B cells, having different immunoglobulin gene rearrangement patterns, make distinct immunoglobulin molecules.
Efficient activation of B cells to secrete immunoglobulin molecules requires interactions with antigen-specific T cells that have become activated during the course of immune response induction. Some of this T-cell “help” is accomplished via secretion of cytokines such as IL-2, but a substantial component is provided by the cell-associated molecule, CD40L, which is essential for directing B cells to generate antigen-specific immunoglobulin G (IgG). Indeed, humans lacking CD40 have a defect in isotype switch, resulting in an accumulation of IgM molecules, with a paucity of IgG and IgA antibody molecules, and a resultant susceptibility to bacterial infection; this condition is known as the hyper IgM syndrome.
Just as molecular characterization of TCR gene patterns can differentiate polyclonal from oligoclonal or monoclonal T-cell populations, immunoglobulin gene rearrangement patterns can also be used to classify B-cell populations since cells present in monoclonal B-cell tumors express identical surface immunoglobulin. The unique structure of a given immunoglobulin’s antigen-binding sequence can be recognized and is known as its idiotype. Identification of a monoclonal population of B cells, all with the same idiotype, allows malignant cells to be distinguished from normal polyclonal B cells, which do not express the same idiotype. Thus, idiotypes can function as tumor-specific markers, and idiotypes have also been the target for individualized immune therapy for some B-cell malignancies. One of the first forms of clinically effective cancer immunotherapy involved generation of unique mAbs against an individual patient’s lymphoma-specific idiotype by Ron Levy and colleagues. More recently, recombinant vaccines are created from the unique idiotype found within the clonal B-cell populations in an individual’s lymphoma, resulting in some effective antitumor effects.20 B cells also have a variety of other immunoregulatory functions that do not derive from their immunoglobulin production capabilities. B cells can serve as
APCs and activators of T cells, and a recently described subset of B cells, known as regulatory B cells or Bregs, have a regulatory function, inhibiting other components of the immune response.
APCs and activators of T cells, and a recently described subset of B cells, known as regulatory B cells or Bregs, have a regulatory function, inhibiting other components of the immune response.
NATURAL KILLER CELLS
NK cells are lymphocytes that do not express mature T- or B-cell markers and are notable for their ability to spontaneously destroy some tumor cells and virally infected populations without requiring prior immunization or activation. This cytolytic capacity is mediated via the same molecules used by cytolytic T cells (i.e., perforin, granzyme, Fas-Fas ligand, and TRAIL-TRAIL receptor interaction, as shown in Fig. 5.6) to induce target cell lysis. Unlike T cells, which require preactivation to induce expression of their perforin- and granzyme-containing granules, NK cells constitutively express cytolytic granules and can kill appropriate targets very rapidly. The granules can be observed histologically and have led to the description of NK cells as large granular lymphocytes. Physiologic roles for NK cells are manifold.21 A primary role for NK cells is first line of defense against viral pathogens, since animals lacking NK function show a great susceptibility to infection with herpes viruses. NK cells also serve as communicators between innate and adaptive immune responses, enhancing the capacity for innate responses to initiate an adaptive response. NK cells also regulate the strength of an appropriate response to infection as evidenced by the fact that individuals with hemophagocytic lymphohistiocytosis (HLH) have a functional deficiency in NK cells. When these individuals are challenged with certain types of infection, they can undergo life-threatening systemic immune activation and cytokine release syndrome. NK cells can also play a direct role in preventing the growth of cancer, through a process described as immune surveillance (discussed below).22
The important role that NK cells play in preventing tumor growth and their potential as therapeutic agents in cancer immunotherapy has recently evolved as a result of a better understanding of the components of NK cell recognition. Unlike T cells, which require MHC expression on target cells to induce cytolysis, NK cell lysis is inhibited by the presence of self-MHC molecules, thereby giving rise to a model for NK cell/target cell interaction, termed the missing self hypothesis. This model holds that NK cells are normally prevented from granule release by “tonic” engagement of inhibitory receptors, with the prototypic inhibitory ligand being self-MHC. NK inhibitory receptors comprise three major families: (a) the killer cell immunoglobulin-like receptors (KIRs), polymorphic molecules characterized in humans containing an immunoglobulin-like domain; (b) the CD94/NKG2 family of lectin-type molecules, also predominantly characterized in humans; and (c) the Ly49 family, which are present in mice and comprise type II transmembrane protein dimers.
An individual’s KIR repertoire is inherited independently from MHC molecules, and contains varying proportions of activating and inhibitory receptors depending on the haplotype. Thus, the degree of natural, endogenous NK reactivity varies substantially between individuals and correlates with the propensity to develop autoimmune diseases and/or the ability to control viral infections.23 Distinct KIR families recognize and prevent killing of cells expressing a specific family of MHC class I molecules. KIR molecules largely recognize two families of HLA-C alleles, with the KIR2DL2/3 family of KIRs binding HLA-C group 1 and the KIR2DL1 family binding HLA-C group 2.24 Diversity in KIR expression in an individual is endowed not only by the genetic content at the KIR locus but also during NK cell development. The KIR expression profile is such that the majority of NK cells express at least one KIR that is inhibited by self-MHC. However, target cells expressing non-self MHC or lacking MHC altogether are capable of killing targets if activating NK receptors are encountered (discussed later). The recognition and inheritance pattern of NK KIRs has recently provided an option for enhancing NK-mediated antitumor effects in the context of MHC-mismatched allogeneic stem cell transplantation. When donor-recipient pairs have disparate KIR expression and HLA-C alleles, the NK cells from the donor can recognize and kill target cells due to the lack of a corresponding inhibitory ligand in the recipient. While one might intuitively expect rampant GVHD as a result of HLA-C mismatch and resultant KIR-mediated NK-cell activation, the pattern of NK attack is largely limited to hematopoietic tissues and tumor tissues. In fact, investigators have shown a beneficial impact of HLA-C mismatch in stem cell transplantation, with NK-mediated alloreactivity diminishing GVHD and providing dramatic graft-versus-leukemia (GVL) effects, especially in myeloid leukemia.24
Current models hold that NK cell lysis is not entirely dependent on the absence of an inhibitory ligand but, rather, represents a balance between inhibitory and activating signals. Thus, the lack of GVHD as a result of NK reactivity in the setting of hematopoietic transplantation has been hypothesized to be due to lack of expression of NK-activating receptor ligands on most normal tissues. Among the numerous activating NK receptors is FcγRIII (CD16), which binds cell-bound IgG molecules recognizing cell surface antigens. Through this interaction, NK cells can recognize, be activated by, and destroy cells that are “opsonized” by antibodies bound to cognate antigen on the cell surface, through a mechanism designated antibody-dependent cell-mediated cytotoxicity (ADCC), which is of central importance for the efficacy of tumor-directed mAb-based therapy. This property has recently served as the basis for integrating IL-2 infusions into treatment regimens with chimeric mAbs, with the goal of enhancing ADCC function via NK cell activation with IL-2.25
Other NK-activating receptors include NKG2D, a C-type lectin receptor found on virtually all human and murine NK cells, which induces potent activation of cytokine release and induction of cell-mediated lysis. NKG2D is activated by tissue-restricted ligands expressed on cells that have been “stressed” (i.e., virally infected cells, cells exposed to “heat shock,” and many types of malignant cells). In humans, the ligands that activate NKG2D are the MHC class I polypeptide-related sequence A and sequence B molecules (designated MICA and MICB). While most epithelial cancers overexpress MICA, high levels of soluble MICA found in the serum of these cancer patients can inhibit NK reactivity. Other activating NK receptors include the NKp44, NKp46, and NKp30 receptors.26 Recent evidence in humans indicates that expression of activating and inhibitory receptors can be modulated during immune reconstitution and, particularly in the setting of viral infections.27,28
A fuller understanding of the biology of this system continues to evolve,29 but the current NK activation model integrates NK recognition of “stressed cells” by activating ligands with the simultaneous requirement for recognition of “missing self.” This model explains why NK cells may preferentially attack stressed or neoplastic cells that have down-modulated MHC molecules. In theory, such a model suggests that NK cells provide an “immune surveillance” mechanism selectively targeting tumor cells or virally infected cells that down-modulate MHC molecules. Concurrently, the T-cell arm of the immune system would be poised to lyse MHC-bearing cells that express foreign antigens recognizable by their TCRs. One could envision a scenario wherein down-modulation of one MHC allele that may be necessary to present a particular tumor-specific peptide may diminish the tumor’s capacity to be recognized by classical T cells, while maintenance of another allele, reactive with the KIR receptors, could turn off NK cell-mediated cytolytic responses. In this way, the tumor could subvert both the T-cell and NK-cell immune response.30
Recently, new approaches for expanding NK cells ex vivo have been developed,31 and IL-15 has been identified as a major growth factor for peripheral NK cell expansion,32 raising new possibilities for therapeutic manipulation of this axis.33 Indeed, since the natural evolution of the immune system involved all components working together, one futuristic vision of immunotherapy would integrate an mAb targeting a cell surface receptor, with a therapy
designed to augment NK-cell number and function to enhance antibody-mediated ADCC and target tumor cells that have down-regulated MHC molecules, with T-cell-based immunotherapy to target MHC-expressing tumor cells that could synergize with NK-cell-mediated cytotoxicity. In preclinical studies, such combined immunotherapies have shown promise.34
designed to augment NK-cell number and function to enhance antibody-mediated ADCC and target tumor cells that have down-regulated MHC molecules, with T-cell-based immunotherapy to target MHC-expressing tumor cells that could synergize with NK-cell-mediated cytotoxicity. In preclinical studies, such combined immunotherapies have shown promise.34
IMMUNE FUNCTION IN CANCER PATIENTS
It is widely accepted that cancer patients have variable degrees of immunosuppression. The causes of cancer-associated immunosuppression are multifactorial and include anatomic alterations of lymphohematopoietic organs by tumors, immunosuppressive mediators released by tumors (e.g., TGF-β, IL-10, and VEGF), malnutrition, physiologic stress, and most importantly, immunosuppressive effects of agents used to treat cancer. Cancer-associated immunosuppression can be clinically significant; combined abnormalities in innate and adaptive immunity, such as those that exist in acute leukemia with pancytopenia, are associated with a high incidence of infectious complications. Patients with solid tumors also show subclinical abnormalities in T-cell immunity at the time of diagnosis, prior to induction of therapy. The most immediate effect of conventional cancer chemotherapy on the immune system is neutrophil depletion, which predisposes patients to bacterial infection (reviewed in Chapter 41). Monocytes are also depleted following chemotherapy, but they recover rapidly, precede neutrophil recovery, and often “overshoot” to supraphysiologic levels. Less is known about quantitative and qualitative effects of chemotherapy on monocyte-derived macrophages, myeloid-derived suppressor cells, and DCs in tissues.35 In murine models, monocyte recovery following cytotoxic chemotherapy induces expansion of myeloid-derived suppressor cells, discussed earlier in the chapter.
Cytotoxic chemotherapy also depletes lymphocyte populations, with the susceptibility and rate of recovery varying among lymphocyte subsets. NK cells are generally resistant to cytotoxic chemotherapy and recovery is rapid and complete. B cells are often profoundly depleted following cytotoxic chemotherapy, and when cycles of chemotherapy are administered over several months, this can result in a complete absence of circulating B cells for the duration of therapy. B-cell depletion results in diminished circulating IgM and IgA levels, whereas plasma IgG levels typically remain more normal since plasma cells are relatively resistant to cytotoxic chemotherapy and irradiation. The impact of IgM and IgA deficiency on infectious complications or antitumor immunity has not been well studied. Cytotoxic chemotherapy can also induce substantial T-cell depletion; the degree of CD4+ depletion generally correlates with the degree of immunosuppression. Because it is difficult to predict how immunosuppressive a particular regimen will be for an individual patient, quantification of lymphocyte populations by using flow cytometry can be useful to estimate the level of immunosuppression induced by cytotoxic chemotherapy, with increasing risk for opportunistic infections when CD4+ counts are less than 200 cells/µL. In practice this is not generally done but in selected high-risk patients it may be something to consider. Persistent T-cell depletion over time provides more compelling evidence for immunosuppression than isolated, transient decreases in peripheral blood T-cell numbers associated with physiologic stress. In a pediatric oncology population, T-cell immunodeficiency is most severe and most common following allogeneic bone marrow transplantation (BMT),36 in which a variety of factors, including GVHD, contribute to a high incidence of opportunistic complications (discussed in Chapters 16 and 40).
Among individual chemotherapy agents, the purine nucleoside analogs (including clofarabine and fludarabine) are the most potent class of lymphocyte-depleting agents. Similarly, the anti-CD52 mAb (Campath) induces severe T-cell depletion. Cyclophosphamide is also commonly associated with chemotherapy-induced immune suppression, with a substantial dose-response effect, such that severe immunosuppressive effects can be seen with doses greater than 3 g/m2. Thus, essentially all patients treated with purine nucleoside analogs, Campath, or high-dose cyclophosphamide, should be considered for pneumocystis prophylaxis.37 More commonly, cyclophosphamide is administered as part of a multiagent regimen where even lower doses can cause significant lymphocyte depletion, increasing the risk of opportunistic complications.
Corticosteroids are commonly administered in the context of cancer therapy as antineoplastic agents, antiemetics, prophylaxis against hypersensitivity reactions, or to control cerebral edema and can predispose cancer patients to fungal, viral, and other opportunistic infections. The immunosuppressive effect of corticosteroid therapy on host immunity is primarily due to dampening of innate immune responses. Mature T cells are relatively resistant to corticosteroid-induced lysis but T cell function can be impaired by exposure to corticosteroids. Corticosteroids inhibit the development of immature myeloid DCs from monocytes, inhibit myeloid DC maturation and cytokine secretion,38,39 and inhibit IFN-α-producing pDCs.
Unlike regeneration of neutrophils, monocytes, and NK cells following chemotherapy, which is predictable and complete in most patients, T-cell regeneration is slow and incomplete, and shows a high level of variability among patients. As a result, T-cell depletion induced by cytotoxic antineoplastic therapy is typically followed by slow and incomplete restoration of peripheral T-cell populations, some of which occurs from hematopoietic stem cells via the more efficient thymic-dependent differentiation pathway and some via thymic-independent expansion of residual mature T cells. It is clear that recovery of CD4+ T-cell populations is highly age-related, especially when comparing children with young adults, because children typically show evidence for brisk recovery of thymopoiesis while adults have delayed or absent thymic recovery. Direct thymic insults, such as GVHD, HIV, or mediastinal irradiation, also substantially diminish thymic reserve, rendering even young patients susceptible to chronic CD4+ depletion.40
For patients without sufficient thymic reserve to rapidly repopulate T cells via thymic-dependent pathways, T-cell regeneration occurs primarily via homeostatic peripheral expansion of residual mature T cells. Homeostatic expansion cannot fully restore T-cell number or TCR repertoire diversity, and homeostatically expanded T cells demonstrate limited functional efficacy. Despite these shortcomings, homeostatically expanded cells undergo exaggerated proliferative responses to strong antigens and can uniquely respond to weak, self-antigens, which are typically ignored by “healthier” T cells. Because tumor antigens are weak self-antigens, lymphopenia has been induced in experimental protocols prior to administration of adoptive T-cell immunotherapy for cancer, so that the augmented reactivity characteristic of homeostatic peripheral expansion can be exploited for antitumor immunotherapeutic benefit.41,42
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


