Chapter Outline
Leukoreduction of Red Blood Cells
Irradiation of Red Blood Cells
Selection of Blood for Transfusion to Patients with Blood Group Alloantibodies
Selection of Blood for Transfusion to Patients with Blood Group Autoantibodies
PRINCIPLES OF HEMATOPOIETIC CELLULAR THERAPY
ERYTHROCYTAPHERESIS (RED BLOOD CELL EXCHANGE) FOR SICKLE CELL DISEASE
PERIPHERAL BLOOD PROGENITOR CELLS
* Revised from a chapter in an earlier edition whose authors included David F. Friedman, Grace Kao, Richard M. Kaufman, and Leslie Silberstein.
Transfusion medicine focuses on the administration of blood, blood components, and purified blood proteins to patients for therapeutic purposes. Red blood cells (RBCs) are the most frequently used blood component. In this chapter RBC transfusions are discussed first, and issues that pertain to all transfusions, such as transfusion reactions and transfusion-transmitted diseases, are discussed in this section. Platelets are often transfused to some patients, such as those receiving oncologic treatment, and are discussed in the next section of this chapter. While not a traditional blood component prepared by blood banks, hematopoietic progenitor cells are a blood derived cellular component that are being infused with increasing frequency over the past two decades. Although these cells are often said to be transplanted rather than transfused, they are also discussed in this chapter. Finally, granulocyte components collected by apheresis for transfusion and other apheresis procedures that allow for the simultaneous removal and transfusion of blood components are discussed at the end of this chapter.Red Blood Cell Transfusions
Indications For Transfusion
The primary function of RBCs is to bind oxygen as the RBCs circulate through the pulmonary bed and release oxygen in the capillaries at a sufficiently high pressure for tissue diffusion. The configuration of the oxyhemoglobin dissociation curve provides for proper diffusion as long as only part of the oxygen is released during capillary transit. The oxygen tension of fully oxygenated blood in the arteries is approximately 100 mm Hg; a release of 20% of the oxygen in the capillaries maintains a pressure of 40 mm Hg, which is sufficient to propel oxygen to all cells within a truncated cone segment around the capillary. RBCs compensate for a loss in oxygen-carrying capacity relatively rapidly by a shift in the oxyhemoglobin dissociation curve to the right, which decreases oxygen’s affinity for hemoglobin (Hb). In addition, cardiac output increases. On a more chronic basis, blood volume, the size of the vascular bed, and the rate of production of RBCs increases. These compensatory mechanisms permit states of mild to moderate anemia without significant symptoms.
For most patients the only indication for RBC transfusion is to provide a patient with sufficient RBCs to prevent or reverse tissue hypoxia caused by insufficient compensation. One large multicenter randomized controlled study of adult critically ill patients is probably applicable to some pediatric patients, especially adolescents. That trial compared a liberal transfusion strategy in which patients received RBC transfusions when their Hb level was below 10 g/dL to a restrictive transfusion strategy in which patients were transfused when their Hb level was below 7 g/dL. Among patients younger than 55 years old, those who were transfused according to the more restrictive strategy had a significantly lower 30-day mortality rate. A similar study of stable critically ill pediatric patients in intensive care units found similar clinical outcomes but fewer RBC transfusions in patients transfused at an Hb concentration at or below 7 g/dL compared with patients transfused at an Hb concentration at or below 9.5 g/dL. In that study patients could be transfused for active bleeding or hemodynamic instability even if their blood Hb concentration had not reached the assigned trigger.
Two prospective randomized controlled studies have compared transfusion strategies for low-weight premature infants. The triggers used for transfusion depended on the oxygen support requirements in both studies and also on the baby’s age in one of the studies. Because neonates often receive multiple aliquots from the same RBC unit, there was no statistical difference in donor exposure between the liberal and restrictive transfusion groups in either study. One study by Bell and colleagues found that neonates on the liberal transfusion strategy were less likely to develop intraventricular hemorrhage or periventricular leukomalacia. However, a follow-up study of these patients when they were between 8 and 15 years old found better neurocognitive development in the children transfused more conservatively. In contrast, the other larger multicenter study by Kirpalani and coworkers found no differences in short-term outcomes between patients assigned to liberal and restrictive groups, but a follow-up study of the patients when they were 18 to 21 months age suggested cognitive delay in those transfused more conservatively. Taken together, these studies suggest that a more liberal transfusion strategy might be advisable. A liberal RBC transfusion protocol might protect against obvious brain injury without significant risk in the neonatal period. Additionally, the larger of the two randomized studies suggested better neurocognitive outcomes with a more liberal use of RBC transfusions.
Children who are chronically anemic as a result of thalassemia may also benefit from a more liberal transfusion policy. These patients benefit from a hypertransfusion program that maintains their Hb concentration above 9 to 10 g/dL. Hypertransfusion of these patients allows them to attain normal height and weight and reduces the risk of hepatosplenomegaly, osteoporosis, and cardiac dilation. Transfusions benefit these patients by increasing tissue oxygenation and particularly by suppressing endogenous erythropoiesis.
Acute Blood Loss
RBC transfusions are frequently used to replace blood loss either in the operating room or after trauma. In practice, diagnosis of the presence and degree of blood loss is quite difficult, especially in a young otherwise healthy child. In fact, a child may sustain a relatively large hemorrhage with few external signs of distress. Signs of impending shock, such as pallor, anxiety, and tachypnea, are frequently subtle and easily overlooked or attributed to other causes. By the time signs of cardiovascular compromise become evident (i.e., stupor, tachycardia or bradycardia, hypotension, coolness of the extremities, weak peripheral pulses, and decreased capillary filling), the patient has most likely lost at least 25% of the total blood volume. Hypotension is one useful clinical sign of moderate to severe blood loss. Patients who have lost more than 25% of their blood volume frequently manifest age-related systolic hypotension: less than 65 mm Hg (younger than 4 years of age); less than 75 mm Hg (5 to 8 years of age); less than 85 mm Hg (9 to 12 years of age); and less than 95 mm Hg (adolescents and adults).
Chronic Transfusion Therapy
In chronic anemia the body is deficient in red cells, whereas the plasma volume is normal or increased so that the total blood volume is normal. Box 36-1 lists the childhood conditions that are likely to require intermittent or long-term transfusion therapy.
Chronic renal failure
Thalassemia
Sickle cell disease
Aplastic anemia
Constitutional
Acquired
Oncologic (leukemia, solid tumors)
Diamond-Blackfan anemia
Transient erythroblastopenia
The following guidelines apply when planning for chronic transfusions (months to years):
- 1.
If possible, transfusion should be avoided or minimized by aggressive treatment of the underlying disorder (e.g., recombinant human erythropoietin [r-HuEPO] administration for patients on dialysis or with chronic renal failure).
- 2.
Leukocyte-reduced RBCs should be used to avert or delay sensitization to leukocyte alloantigens, the most common cause of febrile nonhemolytic transfusion reactions.
- 3.
RBCs should be administered in an amount sufficient to prevent symptomatic anemia and allow for normal growth.
- 4.
The patient’s iron status should be monitored through determination of serum ferritin, iron, iron-binding capacity, and liver iron stores (see Chapter 21 for more information).
Packed Red Blood Cells
A packed cell product is the component of choice for replacement during surgery, RBC loss, and chronic transfusion therapy. Most RBC units contain an additive nutrient solution such as Adsol or Nutricel and have a hematocrit concentration of 50% to 60%, whereas RBC units collected and stored in citratephosphate-dextrose-adenosine (CPDA-1) have an average hematocrit concentration of approximately 75%. Additives vary in their composition and contain some combination compounds, including dextrose, citrate, mannitol, and adenine. Although no systemic pediatric risk has been demonstrated for additives, some institutions avoid using them or remove them before transfusing in large quantities to neonates.
Blood Storage
During storage RBC metabolism continues, and some leakage through RBC membranes occurs. This results in decreasing plasma dextrose and increasing plasma potassium. These changes are acceptable for most transfusions, but in some situations the increased potassium in solution may cause problems. In these cases fresh blood (less than 7 to 10 days old) may be beneficial. This is indicated when small patients receive large amounts of blood, such as neonates receiving exchange transfusions or infants receiving large transfusions rapidly during surgery. However, neonates can tolerate 5 to 20 mL/kg transfusions of older units of RBCs, and some blood banks dedicate a unit of blood for repeated transfusions to one infant to minimize the number of donors to which each infant is exposed. This protocol can lead to transfusion of older RBC units, which are safe for infants, including premature infants.
Leukoreduction of Red Blood Cells
Transfusion of allogeneic cellular blood products is associated with deleterious effects caused by the presence of residual leukocytes. These side effects include alloimmunization to histocompatibility antigens in patients with leukemia, transmission of some viruses, febrile reactions, and graft-versus-host disease (GVHD). Filters are used to reduce the number of leukocytes from whole blood, RBCs, and platelets. Routine leukoreduction of red cells to less than 5 × 10 6 leukocytes per unit reduces the frequency of febrile nonhemolytic transfusion reactions in patients who are transfusion dependent. The removal of leukocytes from blood also reduces the risk of leukocyte-associated virus transmission, such as cytomegalovirus (CMV). Leukoreduction of RBCs and platelets to a level of less than 5 × 10 6 has been demonstrated to substantially reduce CMV transmission in neonates and transplant patients. However, leukoreduction is not sufficient to prevent transfusion-associated GVHD.
Irradiation of Red Blood Cells
Transfusion-associated GVHD is a rare complication after transfusion of cellular products. Patients who are the most susceptible include premature neonates, some patients with congenital immunodeficiency syndromes, immunosuppressed oncology patients being treated with chemotherapy or irradiation (or both), and patients receiving certain immunosuppressive drugs after solid organ transplantation and certain other indications. In addition, normal recipients who are heterozygous for human leukocyte antigens (HLAs) do not reject lymphocytes that are transfused from a donor who is homozygous for one of the recipient’s haplotypes. This is not merely theoretic. Transfusion-associated GVHD has developed in normal recipients undergoing surgery who received unirradiated directed donor blood from a first-order relative. Because of the wide variety of patients at risk for GVHD, some transfusion services have chosen to irradiate all cellular blood products to ensure that no patient develops this life-threatening complication.
Immunologic Considerations
Multiple antigens that are expressed on erythrocyte cell surfaces can be recognized by the immune system of a transfusion recipient. Before an RBC transfusion, erythrocytes that are immunologically compatible with the patient must be identified. Erythrocyte antigens are polymorphic inherited structural characteristics located on proteins, glycoproteins, or glycolipids on the outside surface of the RBC membrane. Erythrocyte antigens are clinically important in the immune destruction of RBCs in allogeneic blood transfusions, maternal-fetal blood group incompatibility, autoimmune hemolytic anemia, and organ transplantation. The ability to detect and identify antibodies to erythrocyte antigens and identify compatible donor RBCs lacking the specific antigens has contributed significantly to the safe supportive blood transfusion practices used today.
The ABO and H System
The H Antigen.
A and B antigens on RBCs are defined by a terminal sugar attached to a carbohydrate chain on membrane glycosphingolipids. The H antigen is the precursor molecule on which A and B antigens are built. The H antigen on RBCs is defined by fucose at the terminus of carbohydrate chains. Expression of the H antigen is determined by a fucosyltransferase that transfers a fucose to an oligosaccharide side chain that is attached to integral membrane proteins on RBCs. The fucosyltransferase (and thus the H antigen) is present in all persons except those with the rare Bombay (Oh) phenotype.
The A and B Antigens.
Transferases responsible for the A or B antigens add N- acetyl galactosamine or galactose, respectively, to the precursor chain carrying the H antigen ( Fig. 36-1 ). The genes for the A and B blood group antigens are codominant because people with both transferases will transfer galactosamine and galactose and thus express A and B antigens. Group O is the amorph of the ABO blood group system. RBCs from group O persons have H antigens but lack A and B antigens.
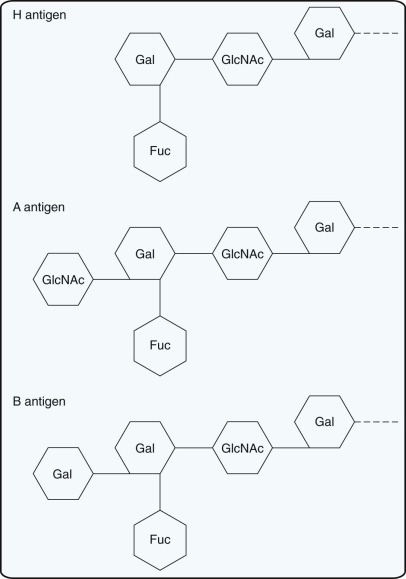
Although ABO antigens are detectable on the RBCs of 5- to 6-week-old embryos, these antigens are not fully developed until 2 to 4 years of age. RBCs from infants carry A, B, and H antigens on linear oligosaccharide chains, which have only one terminus to which the A, B, and H sugars can be added. The fact that the A and B antigens are not fully developed at birth may be one reason that ABO hemolytic disease of the newborn (HDN) is usually a mild disease. As the infants grow older, the oligosaccharides branch, creating additional termini to which A, B, and H sugars can be added. After the children reach 2 to 4 years of age, expression of A and B antigens remains fairly constant throughout life.
Antibodies to A and B antigens, also known as isohemagglutinins, develop as a result of environmental stimulants, such as bacteria, that carry similar antigens. Because isohemagglutinins are naturally occurring, it is essential to select ABO-compatible blood for transfusion. Indeed, ABO incompatibility is the most frequent cause of fatal hemolytic transfusion reactions.
Isohemagglutinin production begins several months after birth, reaching a peak at 5 to 10 years of age and declining with increasing age. Thus infants younger than 4 months of age who have isohemagglutinins will have acquired them through placental transfer of maternal immunoglobulin G (IgG) anti-A, anti-B, or anti-A,B.
Isohemagglutinins from group A and B individuals are predominantly immunoglobulin M (IgM) that do not usually cross the placenta and cause HDN. However, as group O serum contains IgG isohemagglutins, ABO HDN is most frequently seen in non–group O infants of group O mothers.
The Lewis System
The Lewis antigens, Le a and Le b , are not intrinsic to RBCs but are instead carried on plasma glycosphingolipids that are adsorbed onto the RBC membrane. These antigens are structurally related, with the Le b antigen containing an additional fucose that is not present on the Le a antigen. Furthermore, the one additional fucose present on the Le b antigen is not very different from the Le a antigen. Thus Le a individuals rarely make anti-Le b . Hence in most cases Lewis antibodies are made only in individuals who are Le(a−b−).
In most cases Lewis antibodies do not cause hemolysis, and donor RBCs lacking the Lewis antigens do not usually need to be selected. However, rare hemolytic transfusion reactions have been reported when Le a + RBCs were transfused to recipients with anti-Le a . This may occur when the antibody reacts at 37° C. In these cases it is not difficult to identify Le a − donor units because the RBCs in almost 80% of donors are Le a −. Anti-Le a and anti-Le b are not known to cause HDN. They are usually IgM, and fetal RBCs type Le(a−b−). Interestingly, Lewis antigens are also expressed on Helicobacter pylori, a causative agent of gastric ulcers, and play a role in adhesion and colonization of the bacteria in the gastric mucosa.
The Ii Collection
The I and i antigens reside in the subterminal portions of carbohydrate chains on glycolipids or glycoproteins of the RBC membrane. The i antigen on fetal RBCs appears when disaccharide units are linked in a straight chain. In the first 2 years of life, many of these linear chains are modified into branched chains. I specificity results at the expense of i antigens when the branched structures appear. Adults have RBCs with predominantly I antigens and little or no i antigen, whereas newborns have RBCs with strong expression of i antigens and small amounts of I antigen.
Anti-I is a common autoantibody that is reactive at room temperature and below and usually produces no hemolysis. In cold agglutinin disease, anti-I causes in vivo hemolysis, and such pathologic autoantibodies often react at 30° C in albumin. Adult I-negative RBCs are not readily available for transfusion, and patients with cold agglutinin disease caused by anti-I are transfused with I-positive RBCs through a blood warmer. The titer and thermal range of anti-I is often increased after infection with Mycoplasma pneumoniae . The presence of a cold agglutinin caused by an anti-I antibody frequently occurs in M. pneumoniae infection, but this is neither a specific nor a sensitive test for this infection. Analogously, cold agglutinins resulting from anti-i antibodies are sometimes present in Epstein-Barr virus infection, but this is neither a sensitive nor a specific test for this infection.
Levels of the i antigen can aid in differentiating Diamond-Blackfan anemia from transient erythroblastopenia of childhood. The i antigen is enhanced on RBCs from patients with Diamond-Blackfan anemia and is absent or of reduced strength on RBCs from children with transient erythroblastopenia of childhood. However, the enhanced i antigen associated with Diamond-Blackfan anemia reflects stress hematopoiesis and is not a specific aspect of the disease. In transient erythroblastopenia of childhood, the reduced levels of i antigen reflect the selective suppression of erythropoiesis secondary to infection; however, during recovery from this disorder the patient’s marrow is active and the i antigen may be enhanced as a result of hematopoietic stress.
The P System
Like the A, B, and H antigens, the P blood group antigens are defined by sugars added to precursor glycosphingolipids. Antigens in this group include P1, P, and P K . Among white people, 21% have the P1-negative phenotype, and they commonly produce anti-P1. This cold reactive IgM antibody does not cross the placenta and has rarely been reported to cause hemolysis in vivo. An autoantibody, autoanti-P, is found in patients with paroxysmal cold hemoglobinuria and in some patients with acquired immune hemolytic anemia. It is called the Donath-Landsteiner antibody and is most frequently seen in children. This IgG autoantibody is a biphasic hemolysin that binds to RBCs in the cold and then hemolyzes them when warmed. The patient’s serum causes hemolysis of RBCs that have been incubated first in melting ice and then incubated at 37° C. This antibody should be considered when the patient has hemoglobinuria or anemia (or both) and C3 alone is present on the RBCs.
The P antigen serves as a receptor for parvovirus B19 and pyelonephritic Escherichia coli . Persons with the very rare p phenotype lack the antigens P1, P, and P k and are not susceptible to infection by parvovirus B19.
People with the rare p phenotype may produce anti-P1+P+P k , a potent hemolytic antibody that can cause immediate hemolytic transfusion reactions. This antibody has also been associated with HDN, fetal death, and miscarriages during early pregnancy in women whose RBCs have the p phenotype.
The Rh System
The Rh blood group system includes D, C, c, E, e, and 40 other antigens. The terms Rh positive and Rh negative refer to the presence and absence of the D antigen, respectively. The D antigen is, after A and B, the most important RBC antigen in transfusion practice. Persons who lack the D antigen do not have anti-D unless they are exposed to D antigen by pregnancy or transfusion. More than 80% of persons who lack the D antigen can produce anti-D after receiving a unit of D-positive donor blood, but only approximately 22% of D-negative patients transfused with D-positive RBCs make an anti-D antibody. Although these statistics probably do not apply to infants younger than 4 months of age, some infants probably can respond to the Rh(D) antigen.
The RBCs of 0.2% to 1% of white individuals and a greater percentage of African Americans carry reduced expression of the D antigen known as weak D. Some RBCs expressing weak D may not be detected by routine anti-D reagents, but almost all are detected by a more sensitive test, the indirect antiglobulin test. D and weak D have the same RH (D) protein complex on their surface but weak D RBCs have fewer RH (D) proteins. That is usually due to point mutations in the gene encoding the Rh(D) antigen, the RHD gene. Most patients of the weak D phenotype can safely receive blood transfusions from individuals who are Rh(D) positive. However, some individuals express a truncated form of the D protein, known as a partial D, and can make an anti-D antibody. Erythrocytes from such an individual may type as D-positive or weak D. Often only reference labs can determine the specific type of weak D that is present, sometimes using molecular techniques.
All D-negative donor blood is tested for weak D by the antiglobulin test to ensure that D-negative patients are not transfused with weak D RBCs that can induce an anti-D antibody that can hemolyze weak D RBCs. Indeed, there are rare reports of fetuses with weak D having HDN when born to a mother with anti-D antibody in her plasma. Weak D donor RBCs are labeled as Rh(D) positive to ensure that Rh(D)-negative individuals do not receive weak D RBCs. Analogously, at birth an infant is designated as Rh(D) negative only when negative results are obtained for routine anti-D and weak D. This ensures that Rh(D)-negative mothers receive postpartum doses of Rh immune globulin if their baby is Rh(D) positive or weak D positive.
Rh antigens are carried on Rh proteins that pass through the RBC membrane 12 times and are encoded by at least two genes on chromosome 1. These Rh proteins associate with the RHAG protein that is required for cell surface expression of the Rh antigens. D, C, c, E, and e antigens are associated with specific amino acid substitutions on the Rh proteins. Patients with erythrocytes deficient in Rh proteins owing to complex mutations in the Rh locus have a mild spherocytosis and a mildly hemolytic state known as Rh-deficiency syndrome, or Rh null. Mutations of the RHAG gene result in regulatory-type Rh deficiency (Rh null or Rh mod) associated with mild hemolytic anemia with overhydrated hereditary stomatocytosis showing increased permeabiltity to cations. Although the function of the Rh proteins is unknown, they may play a role in ammonium transport.
The MNS System
The M, N, S, s, and U antigens are carried on sialoglycoproteins on the RBC membrane. M and N are encoded by paired allelic forms of the glycophorin A gene (GPA) and are the result of amino acid substitutions at residues 1 and 5 of GPA. Human RBCs type as M+N−, M−N+, or M+N+, representing homozygosity for M, homozygosity for N, and heterozygosity for both genes, respectively. S and s antigens are encoded by alternative forms of the glycophorin B gene (GPB) . GPA and GPB are encoded by homologous genes on chromosome 4q28-q31. GPA binds E. coli and plays a role in malarial invasion of RBCs. RBCs lacking GPA, GPB, or GPC are resistant to invasion by Plasmodium falciparum to varying degrees.
Anti-M and anti-N can be IgG, IgM, or a mixture of both. These antibodies are usually clinically insignificant and are often present in persons who have not been previously transfused or pregnant. Those antibodies that react at 37° C may cause hemolysis of transfused cells. Antibodies to S, s, and U usually occur after stimulation and are capable of causing hemolytic transfusion reactions and HDN.
The Kell System
The Kell system antigens reside on two transmembrane proteins, the Kell protein and the XK protein, that are linked by a single disulfide bond. The Kell protein is a 93-kd glycoprotein that carries more than 20 antigens, and the XK protein is a 440–amino acid protein that carries just one antigen known as Kx.
The major antigen of the Kell blood group system resides on the Kell protein. This antigen is the K antigen, which is present in 9% of white donors. Anti-K is the most common immune RBC antibody, after those in the ABO and Rh systems. However, because more than 90% of donors are K−, it is not difficult to find compatible blood for patients with anti-K antibodies. The molecular basis of antithetical K and k antigens is an amino acid substitution of methionine to threonine at residue 193 of the Kell protein. This allows for prenatal testing for K and k antigens of amniocytes from a mother whose fetus is at risk for HDN owing to anti-K or anti-k. Antibodies to other antigens on the Kell protein, such as Kp a , Kp b , Js a , and Js b , are less common but are also clinically significant. The Kell protein is a member of a large family of zinc-dependent endopeptidases.
The other protein of the Kell system, the XK protein, carries one antigen, the Kx antigen. The XK protein is encoded by a gene on the X chromosome and is a 444–amino acid integral membrane protein that has some features of a membrane transporter. The absence of membrane XK protein, known as the McLeod phenotype, results in diminished expression of the Kell protein on the membrane and reduced expression of all antigens in the Kell blood group system. The blood of patients with the McLeod phenotype often has a variety of abnormalities, including acanthocytotic erythrocytes, hemolytic anemia, some teardrop erythrocytes, and bizarre poikilocytes. Most patients with McLeod syndrome develop a subclinical myopathy associated with an elevated creatine phosphokinase level. The neuropathy may first manifest as areflexia and is later characterized by basal ganglia atrophy causing dystonic or choreiform movements, psychiatric symptoms, and cognitive changes. Most individuals with McLeod syndrome also develop cardiac symptoms, such as dilated cardiomyopathy and arrhythmias.
The XK gene is less than 500 kb beyond the chronic granulomatous disease (CGD) locus on the short arm of the X chromosome (p21.1). Consequently, males with deletions encompassing both loci have both CGD and McLeod syndrome. Patients with CGD and McLeod syndrome usually make antibodies to the XK and Kell proteins if they are transfused with RBCs containing the XK and Kell proteins. This can lead to major problems in transfusing these patients. To avoid this problem, McLeod patients should be transfused with blood of the McLeod phenotype. Ko blood that lacks the Kell protein is not appropriate because such RBCs have increased expression of the Kx antigen that is expressed on the XK protein. However, male subjects with McLeod phenotype without concomitant CGD (non-CGD McLeod) do not make anti-Kx and can be transfused with McLeod or Ko type blood.
The Duffy System
The Duffy antigens are epitopes present on a cell surface glycoprotein. The Duffy glycoprotein is a chemokine receptor that is expressed on many cell types and can bind a variety of chemokines. Although the Duffy chemokine receptor can bind chemokines, its function is unknown and people lacking the receptor on their RBCs or on all cells are totally healthy.
The two most common alleles in people of European or Asian descent are Fy a and Fy b , which differ by a single amino acid substitution. Anti-Fy a and -Fy b have been implicated in transfusion reactions. Anti-Fy a has caused mild HDN, but anti-Fy b has not been implicated. Anti-Fy a is commonly encountered, but because one third of donors are Fy(a-), compatible blood is not difficult to find.
The RBCs of many people of African descent lack the Fya and Fyb antigens (Fy[a−b−]) on their erythrocytes. However, the Fy b allele is usually expressed in lung, colon, and spleen of these individuals. Thus these individuals cannot mount an immune response to the Fy b antigen but can produce anti-Fy a . In West Africa most black people are Fy(a-b-) as a result of natural selection; these RBCs are resistant to Plasmodium vivax malaria because the Duffy protein is a receptor for P. vivax .
The Kidd System
The Kidd gene encodes two antigens, Jk a and Jk b , which represent a single amino acid substitution (Asp280Asn) in the Kidd glycoprotein. These are usually expressed in one of three phenotypes: Jk(a+b−), Jk(a−b+), and Jk(a+b+). The fourth phenotype Jk(a−b−) is exceedingly rare. Antibodies to both Kidd antigens can cause delayed hemolytic transfusion reactions. Because these antibodies are often weak and may become undetectable over time, they may escape detection in the sensitized patient’s serum before transfusion. If the patient is then transfused with antigen-positive RBCs, the anamnestic response causes an increase in the titer of the antibody and hemolysis of the transfused antigen-positive RBCs. Once the antibody is identified, compatible blood is not difficult to find because one fourth of donors are negative for each antigen.
The Kidd glycoprotein is a urea transporter that prevents RBCs from shrinking and swelling as they pass into and out of the high urea concentration of the renal medulla. The Kidd urea transporter also prevents RBCs from carrying urea away from the renal medulla and decreasing the urea-concentrating efficiency of the kidney. Individuals with RBCs with the Jk(a−b−) phenotype are unable to concentrate urine as efficiently as people who have the Kidd glycoprotein.
The Lutheran System
The Lutheran blood group antigens are carried on a cell surface glycoprotein. The Lu a antigen is present on the RBCs of 8% of people, whereas the antithetical antigen, Lu b , is present in more than 99% of random blood samples. Antibodies in this system are variable and are rarely encountered. The glycoprotein carrying these antigens has five extracellular immunoglobulin-superfamily domains that serve as laminin-binding receptors. Lu glycoproteins may be involved in the pathogenesis of sickle cell vaso-occlusive crises and may also be involved in the metastasis of certain types of malignancy.
About 1 person in 5000 inherits a dominantly acting inhibitor, called In(Lu), which partially suppresses expression of Lu a and Lu b such that they are undetectable by standard agglu-tination tests. The Lu(a-b-) phenotype of the dominant type is associated with acanthocytosis but no hemolysis. Patients with the In(Lu) Lu(a-b-) phenotype have abnormally shaped red cells but no hemolysis. The Lu antigens directly interact with spectrin, independently of protein 4.1, which may partially explain the abnormal RBC morphology in some patients with the In(Lu) phenotype. Osmotic fragility of fresh In(Lu) Lu(a−b−) red cells is normal, but during in vitro incubation the cells lose K + and become osmotically resistant.
Other Systems
Other systems are included in Table 36-1 . Antibodies to antigens in these systems are less common than those described previously, and information regarding their clinical significance is summarized in Table 36-2 .
| Blood Group System | Gene Product |
|---|---|
| Carbohydrate Antigens | |
| ABO | Glycosyltransferase |
| P | Glycosyltransferase |
| Lewis | Glycosyltransferase |
| Hh | Glycosyltransferase |
| Protein Antigens | |
| MNS | Glycophorin A, glycophorin B |
| Rh | D polypeptide |
| RHCE polypeptide | |
| CcEe polypeptide | |
| Lutheran | Lutheran glycoprotein |
| Kell | Kell glycoprotein |
| Kx | Xk glycoprotein |
| Duffy | Fy glycoprotein |
| Kidd | Jk glycoprotein |
| Diego | Band 3 (AE1) |
| Yt | Acetylcholinesterase |
| Xg | Xg a glycoprotein |
| Scianna | Sc glycoprotein |
| Dombrock | Glycoprotein (possibly adenosine 5′-diphosphate[ADP]-ribosyltransferase) |
| Colton | Channel-forming integral protein |
| LW | Glycoprotein |
| Chido/Rodgers | C’ component 4 (C4) |
| Gerbich | Glycophorin C, glycophorin D |
| Cromer | CD55 (DAF) |
| Knops | CD35 (CRI) |
| Indian | CD44 |
| Antibody | IgG | IgM | Complement | Transfusion Reaction Binding | HDN * |
| ABO | Some | Most | Common | Mild to severe; immediate | Moderate |
| MNS | |||||
| MN | Some | Some | Rare | Extremely rare | Extremely rare |
| SsU | Most | Some | Rare | Immediate or delayed | Rare |
| P1 | Rare | Most | Rare | None | None |
| RH | Most | Some | No | Immediate or delayed | Mild/severe |
| Lutheran | |||||
| Lu a | Most | Some | No | None | None |
| Lu b | Most | Rare | No | Mild; delayed | Mild/rare |
| Kell | Most | Some | Some | Immediate or delayed | Mild/severe |
| Lewis | |||||
| Le a | Some | Most | Some | Rare; immediate | None |
| Le b | Rare | Most | None | None | |
| Duffy | |||||
| Fy a | Most | Rare | Rare | Delayed | Mild |
| Fy b | Most | No | Mild; delayed | None | |
| Kidd | Most | Few | Common | Immediate or delayed | Mild/rare |
| Diego | Most | Delayed | Mild/severe | ||
| Yt | Most | Some; mild | None | ||
| Xg | Most | Some | None | None | |
| Scianna | Most | None | None | ||
| Dombrock | Most | Delayed | Mild | ||
| Colton | Most | Mild; delayed | Mild/rare | ||
| LW | Most | Mild; delayed | Mild | ||
| Ch/RG | Most | None | None | ||
| H | Rare | Most | None (except in O h ) | None | |
| XK | Most | None known | Delayed | None | |
| Gerbich | Most | Some | Some | Immediate or delayed | None |
| Cromer | Most | Delayed | Mild | ||
| Knops | Most | None | None | ||
| Indian | Most | None | None | ||
* The most severe form of hemolytic disease of the newborn (HDN) that has been reported is indicated. Remember that exceptions exist.
Maturation of Blood Group Antigens
Several blood group antigens are not expressed or are only weakly expressed on cord RBCs and usually reach adult levels by 2 years of age. Antibodies to these antigens are unlikely to cause HDN. Cord RBCs do not express Le a , Sd a , Ch, Rg, or AnWj antigens. Cord RBCs express the following antigens more weakly than RBCs from adults: A, B, H, I, Le b , P 1 , Lu a (but not Lu b ), Yt a , Vel, Do a , Do b , Gy a , Hy, Jo a , Xg a , and Bg.
Clinical Significance of Blood Group Antibodies
A blood group antigen has no immediate untoward effect on blood transfusion. It is the corresponding antibody that has clinical relevance and dictates the need for tests in a transfusion service to detect blood group antibodies.
Antibodies recognizing antigens in the ABO blood group system are by far the most clinically significant. With antibodies in the other blood group systems, as a general rule, the more immunogenic an antigen, the more clinically significant its corresponding alloantibody. Other clinically significant antibodies occur in the following order, from most commonly to least commonly encountered in transfusion practice: anti-D, anti-K, anti-E, anti-c, anti-Fy a , anti-C, anti-Jk a , anti-S, anti-Jk b . All other clinically significant antibodies occur with an incidence of less than 1% of serum containing antibodies. Anti-P 1 , anti-M, anti-N, anti-Lu a (Lutheran), anti-Le a , anti-Le b , and anti-Sd a are all considered clinically insignificant unless the alloantibodies are reactive in tests performed strictly at 37° C. Clinically insignificant antibodies that react at 37°C by indirect antiglobulin test are those of the Knops and Ch/Rg systems and anti-JMH. Other alloantibodies are clinically significant in some patients but not in others; examples are anti-Vel, anti-Ge (Gerbich), anti-Yt a , and anti-Co a (Colton). Table 36-2 summarizes the clinical significance of antibodies to RBC blood group antigens.
The clinical significance of an antibody to a low-incidence antigen depends on whether it was found during crossmatching or prenatal testing. The specificity of a low-incidence antibody detected during compatibility testing need not be identified to locate antigen-negative blood with which to transfuse that patient because the full crossmatch will be positive in the rare event that a unit of donor blood from the stock supply carries the low-incidence antigen. In contrast, if an antibody to an uncommon antigen is detected in the serum of a pregnant woman, identification of the antibody, or determination of the antigen carried by the fetal or paternal RBCs, can be used as a predictor for the likelihood and severity of HDN. Blood for exchange transfusion will not be difficult to find.
If a patient’s serum contains alloantibodies to a high-incidence antigen, blood may be difficult to find. Examples are anti-U, anti-Kp b , anti-Js b , anti-Lu b , and all antigens in the high series recognized by the ISBT Working Party. Whether the investigation is for transfusion purposes or prediction of HDN, the antibody should be identified. This will aid in both the assessment of its clinical significance and the location of appropriate blood for transfusion.
Blood group antibodies in donor plasma present little hazard to an antigen-positive recipient or RBC units that have low plasma volumes. However, components that have significant plasma, such as fresh frozen plasma (FFP) or platelets, should generally be selected to lack ABO antibodies against recipient antigens. If platelets lacking ABO antibodies against the patient are unavailable, there is a small risk of a potentially severe reaction, and some blood banks have implemented procedures to minimize transfusion of incompatible isohemagglutinins. Donor blood is always tested for the presence of antibodies to antigens other than ABO and blood components containing such antibodies are generally not transfused to patients expressing the corresponding antigen.
Sera from some patients contain autoantibodies that recognize antigenic determinants on RBCs from the majority of individuals as well as their own. In such cases it may be impossible to locate compatible blood and only incompatible blood will be available. This may arise in such cases as autoimmune hemolytic anemia, cold agglutinin disease, and paroxysmal cold hemoglobinuria; these are discussed later in this chapter.
Testing Donor Blood
Donor blood is routinely tested for its ABO and Rh blood group status. More extensive typings for other antigens are performed either when antigen-negative RBCs are required for a recipient whose serum contains clinically significant antibodies or when building an inventory of donors with certain antigenic profiles for future use, such as in the transfusion management of sickle cell disease (discussed later). The more alloantibodies are present, especially to high-frequency antigens, the longer the time needed to locate compatible units.
Compatibility Procedures
The routine approaches used involve testing a blood sample from a prospective recipient for ABO and Rh blood groups and for detection of blood group antibodies. In most cases no unexpected antibodies are detected and donor RBCs of the appropriate ABO and Rh blood type will be selected for transfusion. A sample of this blood may be crossmatched with the patient’s serum by an immediate spin procedure or an “electronic crossmatch” in which a validated computer system verifies that the patient is receiving an RBC unit that is ABO and Rh(D) compatible with the patient’s blood group.
When an antibody is detected, an attempt should be made to identify its specificity. Knowing the specificity of an antibody allows the clinician to establish whether it is likely to be clinically significant and the approach that will be necessary to provide an adequate supply of compatible RBCs.
Compatibility in ABO and Rh
Determination of the ABO and Rh(D) type of the patient is the first step in compatibility testing. In neonates the ABO group is determined by testing the neonate’s RBCs only because the plasma may contain maternal antibodies and young neonates’ immune systems do not make isohemagglutinins. In patients older than 4 months, the ABO group is determined by testing both the patient’s RBCs (forward group) and the plasma or serum (reverse group). The forward and reverse groups should correspond, except that infants younger than 9 months old may not yet make isohemagglutinins. In all other patients, until any discrepancy is resolved, only group O RBCs and AB plasma should be given.
Blood and blood components must be selected to be ABO and Rh compatible ( Table 36-3 ). In general, for patients older than 4 months of age, type-specific RBCs and whole blood are selected, which means that donor and recipient have the same ABO and Rh type; it is acceptable to transfuse ABO and Rh compatible but not identical RBCs. For transfusions of a fetus, group O Rh(D−), irradiated, CMV antibody–negative or leukoreduced RBCs (or both) are usually given regardless of the ABO group of the recipient. For neonates some centers transfuse type-specific RBCs, whereas other centers transfuse group O RBCs to all infants. If the RBCs selected for transfusion are not group O, the neonate’s serum or plasma should be tested for maternal anti-A or anti-B IgG antibodies in addition to IgM isohemagglutinins. If anti-A or anti-B is detected, RBCs that are ABO-compatible with the antibodies should be transfused.
| Patients ABO/D | O Positive | O Negative | A Positive | A Negative | B Positive | B Negative | AB Positive | AB Negative |
|---|---|---|---|---|---|---|---|---|
| O positive | + | + | ||||||
| O negative | + | |||||||
| A positive | + | + | + | + | ||||
| A negative | + | + | ||||||
| B positive | + | + | + | + | ||||
| B negative | + | + | ||||||
| AB positive | + | + | + | + | + | + | + | + |
| AB negative | + | + | + | + |
Compatibility in Other Blood Groups
Compatibility testing for blood groups other than ABO is achieved by the antibody screen and crossmatch ( Figs. 36-2 and 36-3 ). These tests detect unexpected RBC antibodies in the patient’s serum. The patient’s serum or plasma—or in the case of a neonate, maternal serum or plasma—may be used for the antibody screen. In the antibody screen the patient’s serum is mixed with group O reagent RBCs, which have most of the important blood group antigens. Because neonates are unlikely to produce new antibodies, the antibody screen must be performed only once per admission if the initial antibody screen is negative. In the crossmatch the patient’s serum is mixed with donor RBCs or “electronically” crossmatched using a validated computer system. In patients older than 4 months of age, the crossmatch is performed for every unit of RBCs or whole blood transfused. In neonates if the initial antibody screen is negative, it is unnecessary to crossmatch donor RBCs.
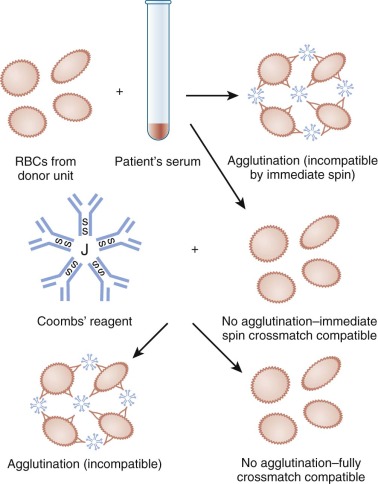
If a blood group alloantibody is detected in the recipient’s serum, every attempt should be made to identify the antibody. The clinical significance of an antibody is based on knowledge of the specificity of the alloantibody and the reported experience in other patients whose serum contains the same antibody specificity (see Table 36-2 ). If the antibody has potential clinical significance, antigen-negative blood should be selected and crossmatched by a procedure that includes the antiglobulin phase.
Selection of Blood for Transfusion to Patients with Blood Group Alloantibodies
Many alloantibodies detected in vitro in human sera do not have the potential to induce in vivo destruction of incompatible RBCs. No technical procedure is able to differentiate, with certainty, between clinically benign and clinically significant antibodies. Although the in vitro characteristics of an antibody may not reflect its behavior in vivo, the following features can be used to help establish its potential significance in a patient. In general, antibodies of the IgG class are more clinically significant than those of the IgM class. Also, the higher the titer of an antibody, the more likely it is to destroy antigen-positive RBCs in vivo . An antibody that causes in vitro hemolysis owing to complement activation (see Table 36-2 ) of antigen-positive RBCs is likely to be clinically significant. Antibodies that react at 37° C are more likely to be clinically significant than those that do not. For cold-reacting IgM antibodies, the wider the thermal range of reactivity in vitro, the more likely it is to be clinically significant. Alloantibodies that react in the cold and are not usually clinically significant include antibodies to M, N, P 1 , Le a , and Le b antigens. In difficult cases a few reference laboratories can also perform a monocyte monolayer assay that has some value in predicting whether an antibody will cause a hemolytic transfusion reaction.
Once a patient is actively immunized to an RBC antigen and produces a clinically significant alloantibody, the patient is considered immunized for life and must be transfused with RBCs lacking the corresponding antigen for life. Neonates with passively acquired maternal antibody are not actively immunized and thus need to be transfused with antigen-negative RBCs only while the antibody is still present.
Emergency Need
When RBC transfusion is urgently needed in a rapidly bleeding patient, compatibility testing should be abbreviated so as not to delay transfusion. The clinician should indicate the need for emergency release of blood and send a pretransfusion patient blood sample to the blood bank. The blood bank can issue group O Rh(D−) RBCs and AB plasma or, if time permits, ABO type-specific RBCs. In some cases of inventory shortages, these patients, especially male ones, may receive Rh(D+) RBCs. In cases of emergency transfusion, the antibody screen and crossmatch may be performed after the blood has been transfused, and the clinician is notified of an incompatible crossmatch.
Autologous Blood
Autologous blood transfusion is possible for some children who undergo elective surgery for which transfusion is anticipated. Transfusion of such blood reduces the risk of transfusion-transmitted viral diseases, but no blood transfusion is without risk. For example, autologous blood transfusions can be contaminated with bacteria or can cause volume overload, and there is always the risk of transfusing the incorrect unit. Intraoperative RBC salvage can be used if tumor cells or bacteria are not expected to contaminate the surgical field.
To donate autologous blood before surgery, a child must meet size or age requirements of the blood donor center, have adequate venous access, and have an Hb concentration of 11 g/dl or greater and a hematocrit of 33% or greater. Some blood donor centers will collect small units that are proportional to the child’s size. Donation should begin 4 to 5 weeks before surgery to allow time for in vivo RBC regeneration. Oral iron supplementation is often given. Recombinant erythropoietin is not routinely recommended for autologous blood donors and has not been licensed by the Food and Drug Administration for autologous donation, but it may be useful in certain circumstances.
Directed Donations
Some institutions allow family and friends to direct their blood donations to be used for a child or infant. This is no safer than random banked blood and adds additional administrative expenses. GVHD can occur when the patient shares an HLA haplotype with an HLA homozygous blood donor, and this is more likely to occur if a patient receives blood from a relative. Hence directed donations from blood relatives should be irradiated, and most institutions irradiate all blood donated by directed donors. Theoretically, blood from some female relatives has a higher chance of containing anti-HLA or antineutrophil antibodies that can contribute to transfusion-related acute lung injury (TRALI). Additionally, transfusion from a relative may induce antibodies to that relative’s HLA antigens, complicating future donations of other tissues or organs from that relative.
Choice of Red Blood Cell Units for the Fetus
For intrauterine transfusions, group O Rh(D−) (and antigen-negative if mother has an alloantibody) RBCs should be used. To prevent graft-versus-fetus disease, blood is irradiated. To prevent CMV infection, CMV antibody–negative RBCs or leukocyte-reduced RBCs (or both) should be used.
Choice of Blood for the Neonate
Cellular components should be selected from CMV antibody–negative donors for neonate recipients weighing less than 1200 g at birth if their mother’s are CMV antibody–negative. Alternatively, leukocyte-reduced blood can also be used to prevent CMV infection if the blood product has less than 5 × 10 6 donor leukocytes . Cellular blood components should be irradiated to prevent GVHD in these low-weight premature infants.
In exchange transfusions for the neonate with HDN, RBCs used must be negative for the implicated antibody (e.g., Rh[D−] RBCs when hemolysis is caused by anti-Rh[D]). In terms of selection of ABO group, group O RBCs and plasma of the infant’s ABO group are often used. Group-compatible RBCs and plasma can also be used if the RBCs are also compatible with any maternal antibodies that are present in the infant’s circulation.
Choice of Blood for Patients with Sickle Cell Anemia
Two important principles must be followed in the transfusion of patients with sickle cell anemia: (1) avoidance of excessive viscosity and (2) prevention of sensitization to RBC antigens.
Sickle cells are intrinsically less deformable than normal cells; thus elevating the hematocrit concentration without significantly reducing the proportion of sickle cells may raise the blood viscosity to dangerous levels. Simple transfusion must be used with caution, particularly in children with hematocrit concentrations greater than 30%.
The question of what to use to transfuse patients with sickle cell anemia has been the subject of many heated debates. There is still no consensus as to the best and most practical approach. Patients with sickle cell anemia, like all patients, are at risk for developing alloantibodies, and alloantibodies may pose specific hazards to patients with sickle cell disease. Patients with sickle cell disease usually need many transfusions throughout their lives, and an antibody to a blood group alloantigen that develops in childhood may be undetectable later in their lives. If they are then transfused with blood expressing that antigen, they are likely to develop an anamnestic response to the antigen and develop a delayed hemolytic transfusion reaction. Patients with sickle cell anemia have been reported to develop painful crises secondary to delayed hemolytic transfusion reactions. Additionally, a patient with sickle cell anemia who develops RBC alloantibodies may be at increased risk for the development of RBC autoantibodies that can significantly complicate the patient’s disease. For these reasons some transfusion services provide phenotype-matched blood for patients with sickle cell disease. Approaches that are used include those described in Table 36-4 . Note that none of these approaches will prevent the production of antibodies to high-incidence antigens, such as U, Js b , Cr a , hr S , and hr B . African Americans tend to be null for antigens commonly expressed in the general donor population, which is mostly Caucasian in the United States. For this reason some blood donor facilities have programs to recruit African Americans to donate blood to support the transfusion needs of patients with sickle cell anemia. One potential risk of this approach is that patients are then likely to be exposed to many more Rh variant genes that are prevalent in the African-American population.
| Approach | Advantages | Disadvantages |
|---|---|---|
| Treat like other patients (i.e., avoid antigens to which the patient has made antibodies) | Simple; avoids unnecessary work | Some patients will make antibodies that can result in serious transfusion reactions that may be especially dangerous in these patients |
| Patients are probably at increased risk for the development of autoantibodies | ||
| Provide fully antigen-matched blood before the patient makes antibodies (i.e., matched for D, C, E, c, e, K, Fy a , Fy b , Jk a , and Jk b antigens). Note that antigen matching for Fy b is not needed for patients of African descent who are Fy(a− b−) because they have the FYB gene, which is expressed in the lung and colon, and do not mount an immune response to the Fy b antigen on transfusion of Fy(b+) RBCs | Reduces the risk of alloantibody and autoantibody formation and the risks associated with these antibodies | Significant work to reduce the risk for an antibody that may never develop; may be difficult to identify units with extended phenotype matches |
| Provide antigen-matched blood after the first antibody is made | Reduces the chance of multiple antibodies with less work than extended phenotype matching for all patients | Some patients will make multiple antibodies quickly |
| Provide partially antigen-matched blood (i.e., matched for D, C, E, and K antigens) | Reduces the chance of multiple antibodies with less work than extended phenotype matching that includes antigens that are not very immunogenic | Some patients will make antibodies against unmatched antigens |
Thalassemia
In thalassemia the goals of transfusion therapy are the prevention of anemia and the maintenance of a circulating Hb level that is sufficient to suppress endogenous erythropoiesis. For most patients maintenance of a pretransfusion Hb level of 9 to 10 g/100 mL is sufficient. RBC alloantibodies are not a significant problem in most patients with thalassemia, possibly because their transfusions are usually initiated at an early age when tolerance may develop or because they don’t generally develop crises associated with hemolysis, as occurs in sickle cell disease. Therefore most institutions in the United States do not provide RBCs that are phenotypically matched to the patient’s red cells.
Choice of Blood for Allogeneic Hematopoietic Progenitor Cell Transplants
Because an HLA-identical donor is usually required for allogeneic hematopoietic progenitor cell (HPC) transplantation, the donor may not have the same ABO and Rh type as the patient. In major ABO incompatibility, the recipient has antibodies against donor RBCs (e.g., recipient is group O and has anti-A and anti-B; donor is group A). In minor ABO incompatibility, the donor has antibodies against recipient RBCs (e.g., donor is group O and has anti-A and anti-B; recipient is group A). These incompatibilities may produce hemolysis, but hemolysis is often minimized by proper selection of blood products for transfusion.
In major ABO incompatibility (e.g., donor is group A, recipient is group O), host-versus-graft reactions can cause delayed erythropoiesis and even RBC aplasia. To prevent recipient destruction of donor RBCs at the time of HPC infusion, donor RBCs are removed from the HPC before infusion. The group O recipient may continue to produce anti-A and anti-B after transplantation and cause hemolysis of the newly produced donor group A RBCs. If transfusions are necessary during this period, group O RBCs can be given ( Table 36-5 ). Before transplantation the group O recipient has anti-A and anti-B. After engraftment of group A donor marrow, the anti-B will persist but the anti-A will become undetectable. Donor group A RBCs are given only after the recipient stops producing anti-A—that is, when the recipient’s group O blood type changes to the donor group A and anti-A is not detected in the patient’s serum.
| Recipient | Donor | Red Cells |
|---|---|---|
| A | O | O |
| A | B | O |
| A | AB | A or O |
| B | O | O |
| B | A | O |
| B | AB | B or O |
| AB | O | O |
| AB | B | B or O |
| AB | A | A or O |
| O | A | O |
| O | B | O |
| O | AB | O |
In minor ABO incompatibility, hemolysis of recipient RBCs by donor antibody is usually preventable. To prevent hemolysis of recipient RBCs by passively transfused donor antibody during HPC infusion, plasma is removed from the HPC preparation before infusion. In addition, passenger B lymphocytes in the donor HPC may produce antibodies that may destroy recipient RBCs. To prevent hemolysis of transfused RBCs, group O RBCs are used for transfusions during the posttransplant period if the antibody is detected. If the donor or recipient has a clinically significant antibody to an erythrocyte antigen, blood lacking the antigen should be given after transplantation.
Choice of Blood for Organ Transplantation
The ABO antigens are strong histocompatibility antigens and thus are of major importance in organ transplantation. Because the ABO antigens are expressed on the vascular endothelium, the recipient can reject and damage vascular endothelium of a donor organ that is a major ABO mismatch. For this reason, major ABO-incompatible vascular organ (liver, kidney, heart, lungs, pancreas) transplants are usually not performed. However, ABO-incompatible heart transplants may be performed in infants and young children who have not yet produced isohemagglutinins or who have produced only low levels of isohemagglutinins.
When organ transplants are performed in the presence of minor ABO incompatibility, the same hemolytic problems as in HPC transplant may occur. Passenger B lymphocytes may produce antibodies against recipient RBCs. Hemolysis of transfused RBCs can usually be prevented when an antibody is detected, by transfusion of group O RBCs for ABO incompatibility and transfusion of antigen-negative RBCs for incompatibility owing to other blood groups.
Selection of Blood for Transfusion to Patients with Blood Group Autoantibodies
Autoimmune hemolytic anemia is described in detail in Chapter 13 . In hemolytic anemia resulting from warm-reactive autoantibodies, no compatible blood can usually be found. The direct antibody test ([DAT]; Fig. 36-4 ) will almost always be positive in autoimmune hemolytic anemia. Blood bank serologic testing is aimed at determining whether underlying clinically significant alloantibodies are present and identifying the specificity of any such antibodies that exist. This can be determined, with time, at a hospital transfusion service, which may require that the testing be performed at an immunohematology reference laboratory. Patients without a history of transfusion or pregnancy are unlikely to have underlying alloantibodies. Transfused RBC units should be compatible with any underlying clinically significant alloantibodies. However, the blood is still usually incompatible with the patient’s serum, and the clinically necessary amount of RBCs should be transfused with careful monitoring but without delay. In some patients the warm autoantibody has a clear-cut specificity for an antigen, and antigen-negative RBCs may be compatible in the crossmatch test.
In paroxysmal cold hemoglobinuria associated with the Donath-Landsteiner antibody, transfusion is not usually necessary. The antibody binds RBCs only at cold temperatures (<20° C). In theory, it is reasonable to transfuse blood through a blood warmer; however, in practice crossmatch-compatible, unwarmed random RBCs have been given without problem. Only rarely is transfusion of RBCs of the rare phenotype p needed. Most cases of paroxysmal cold hemoglobinuria in children are transient and occur after viral infections.
In cold agglutinin disease, a rare condition in children, RBCs should be transfused through a blood warmer. As described previously, the same issue of testing for underlying alloantibodies pertains here.
Adverse Effects of Transfusion
All reactions other than mild allergic (urticarial) reactions require the immediate cessation of the transfusion and sending of a fresh blood sample from the patient to the blood bank for serologic evaluation.
Allergic Reactions
The typical urticarial reaction is characterized by itching and hives. Localized urticaria simply requires interrupting the transfusion and administering an antihistamine. If the symptoms resolve within 30 minutes the transfusion can be continued. Recurrent allergic reactions may require pretreatment with antihistamine.
Some children exhibit more severe allergic reactions, such as laryngospasm and bronchospasm. These patients may require steroids, epinephrine, or both ( Table 36-6 ). The same unit of blood should not be restarted, even after symptoms have responded to treatment. Saline-washed blood products may be helpful in patients with severe or recurrent allergic reactions. Anaphylactic reactions may occur if the patient is IgA deficient and has formed an IgE anti-IgA antibody. In these cases, either washed blood products or products drawn from rare IgA-deficient donors are indicated.
| Agent | Use | Dose | Comment |
|---|---|---|---|
| Diphenhydramine | Treating pruritus and rash (hives) | 6.25 to 25 mg IV over period of 10 to 20 min | Diphenhydramine cannot prevent and has no place in the treatment of severe transfusion reactions |
| Epinephrine | Severe reactions characterized by bronchospasm, hypotension, and shock | 0.1-0.4 mg SC (or IV if the patient is hypotensive) | For patients in shock not responding to epinephrine, treatment with an initial fluid bolus should be initiated; as in patients with shock from other causes, fluids, vasopressors, airway protection, and oxygenation should also be used |
| 1 : 20 Aqueous suspension of epinephrine | After stabilization with epinephrine | 0.05-0.2 mg SC | |
| Fluids | For hypotensive patients, a bolus of 20 mL/kg of normal saline should be administered simultaneously with epinephrine and steroids | ||
| Narcotics | Specific and effective treatment of rigors | 0.1 mg/kg of morphine IV (or equivalent dose of meperidine hydrochloride) | |
| Acetaminophen (Tylenol) | Prevention or reversal of elevations in temperature in mild to moderate febrile reactions | Appropriate for age | |
| Steroids | In moderate to severe reactions; may occasionally be required for severe urticaria; indicated in all reactions characterized by fever, shaking chills, diaphoresis, and pallor | 1 to 2 mg/kg of methylprednisolone (or equivalent dose of dexamethasone or hydrocortisone by intravenous push) |
Febrile Nonhemolytic Reactions
The occurrence of fever, chills, or diaphoresis usually results from cytokines and other pyrogens released from leukocytes and platelets. Cytokines can be released from leukocytes during storage or on infusion to a patient who has antibodies that react with antigens on foreign white blood cells. Febrile nonhemolytic transfusion reactions are not associated with serious sequelae and may be treated with antipyretics. Of note, fever during a transfusion may represent another, more serious types of transfusion reaction: acute hemolysis, sepsis from a bacterially contaminated unit, or TRALI.
Acute Hemolytic Transfusion Reactions
Acute hemolytic transfusion reactions are most often the result of donor-recipient ABO incompatibility. These reactions are generally caused by managerial errors, such as the misidentification of a transfusion recipient. The clinical presentation of acute hemolytic transfusion reactions varies. Signs and symptoms may include any or all of the following: fever, chills, dyspnea, tachycardia, hypotension, flank pain, abdominal pain, pain at the infusion site, apprehension, nausea, vomiting, and hemoglobinuria. Other serious sequelae may include acute renal failure, shock, and disseminated intravascular coagulation (DIC), which may present as bleeding. Some acute hemolytic reactions are characterized by only minor initial symptoms. In anesthetized recipients the only manifestations may be diffuse bleeding (owing to DIC) or red urine (hemoglobinuria). When an acute hemolytic transfusion reaction is suspected, the transfusion should be stopped immediately and an investigation should be initiated. The identity of the patient and unit should be reconfirmed. The remainder of the unit and a fresh blood sample should be sent to the blood bank, where the required workup includes the following: (1) a clerical check, (2) visual inspection of the recipient’s plasma for hemolysis, and (3) a DAT. If there is no hemolysis in the patient’s plasma or urine and the DAT result is negative, immune-mediated hemolysis can be excluded.
Therapy of an acute hemolytic reaction is supportive. Depending on the size of the child, a urine output of 40 to 100 mL/h should be maintained. Early consultation with nephrologists is warranted. Intravenous administration of furosemide or mannitol, which increases renal blood flow, may be required.
Delayed Hemolytic Transfusion Reactions
Delayed hemolytic transfusion reactions are of two different types: primary immunization and anamnestic response. Primary immunization is mild and occurs weeks after transfusion. It rarely causes significant hemolysis and may be suspected after an unexplained decrease in Hb level 2 to 3 weeks after transfusion.
Secondary immune response s occur in patients who have been sensitized to one or more non-ABO blood group antigens during a prior transfusion or pregnancy. When a patient is reexposed to the antigen, an anamnestic response occurs, resulting in a rapid rise in antibody level and accelerated clearance of the transfused antigen-positive RBCs. These patients experience some of the symptoms of a hemolytic reaction 3 to 10 days after transfusion. The clinical syndrome is usually mild, but delayed transfusion reactions can result in profound anemia. These reactions should be suspected in patients who have received multiple transfusions and develop unexplained anemia or bilirubinemia (or both) 3 to 10 days after transfusion. The diagnosis is confirmed by a positive DAT result and identification of RBC alloantibodies in the patient’s serum or on the patient’s RBCs (or both).
Transfusion-Associated Acute Lung Injury
TRALI presents with some combination of fever, chills, dyspnea, cyanosis, hypotension, and bilateral pulmonary infiltrates. The reported incidence is 1 in 5000 transfusions but may be lower in pediatric patients. Many cases appear to result at least in part from antibodies in donor plasma that react with patient HLA or neutrophil antigens. Blood banks minimized the use of plasma from female subjects that is most likely to contain such antibodies, resulting in a significant reduction in TRALI incidence. TRALI is often self-limited, with resolution in 48 to 96 hours in most patients who receive supportive care such as supplemental oxygen and often mechanical ventilation. However, in approximately 20% of patients TRALI can be severe and recovery prolonged; some cases, in fact, are fatal.
Transfusion-Associated Circulatory Overload
Because blood components have high oncotic pressure, transfusion-associated circulatory overload (TACO) may occur, resulting in pulmonary edema. This is mostly seen in adults, although very young children may also be at risk. TACO is treated with diuretics, phlebotomy, and supplemental oxygen.
Transfusion-Associated Gut Injury
Several case series reports have found an association between RBC transfusions and the development of necrotizing enterocolitis (NEC) in premature neonates. Although these studies suggest an association, they do not prove that NEC is caused by transfusion and not by some other factors that occur around the time of RBC transfusions.
Transfusion-Transmitted Diseases
Because of improvements in both donor screening and product testing, blood products are currently extremely safe. But even though the risks of transfusion-transmitted disease have been greatly reduced, they have not been eliminated entirely. Currently, all blood donations in the United States are tested for the presence of human immunodeficiency virus 1 and 2 (HIV-1 and HIV-2), hepatitis B virus (HBV), hepatitis C virus (HCV), human T-cell lymphotropic virus 1 and 2 (HTLV-1 and HTLV-2), West Nile virus (WNV), and syphilis, and all blood donors are tested for Chagas disease at least once. The battery of tests performed includes a combination of serologic tests and nucleic acid tests (NATs) that taken together are exquisitely sensitive. However, very recent infections may be missed (so-called window-period donations). The risks listed in the subsequent sections and summarized in Table 36-7 are based on mathematical models estimating the number of infectious donors whose blood tests negative for a disease because they are in the window period.
| Agent | Number of Infections/Million Units |
|---|---|
| HIV | 0.5 |
| HCV | 0.5 |
| HBV | 2 to 5 |
| HTLV | 0.5 to 4 |
Posttransfusion Hepatitis
Hepatitis B Virus.
The estimated risk of posttransfusion HBV infection is presently 1 in 200,000 to 1 in 500,000 units transfused. Acute HBV infection is symptomatic in approximately 50% of adults, and 5% to 10% of those infected become chronic carriers. In contrast, HBV infection acquired in early childhood is usually asymptomatic and leads to a chronic-carrier state in more than 70% of those infected.
Hepatitis C Virus.
With the routine use of NAT screening, the per-unit risk of HCV is currently estimated to be about 1 in 2,000,000. Children who receive HCV-infected blood products have a 30% to 50% chance of spontaneously clearing their infection. Most others become chronically infected. Approximately 20% of children who have been infected with HCV develop signs of cirrhosis after 18 years of age.
Human Immunodeficiency Virus.
All units are tested for HIV-1 and HIV-2 by immunoassay as well as by NAT. With NAT the window period for HIV has been reduced to approximately 10 days. The per-unit risk of HIV infection is currently estimated to be 1 in 2,000,000.
Human T-Cell Lymphotrophic Virus.
HTLV is the causative agent for both adult T-cell leukemia and endemic spastic myelopathy. Most cases have been reported in residents of or emigrants from Japan, Africa, and the Caribbean region. The most comprehensive report of transfusion-transmitted HTLV indicated that 65% of patients who received whole blood or cellular products from HTLV antibody–positive donors seroconverted. Blood from all donations is tested for HTLV-1 and HTLV-2; the current per-unit risk is estimated to be 1 in 641,000.
West Nile Virus.
WNV is a flavivirus common in Africa, West Asia, and the Middle East. Approximately 20% of infected individuals will develop a febrile illness. Rarely, WNV infection causes an encephalitis syndrome; elderly and immunocompromised individuals are those primarily at risk. WNV has recently become established in North America. The vast majority of cases are transmitted by mosquito bites, but transfusion transmission does occur in rare cases. As with HIV and HCV, NAT has almost completely eliminated WNV from the U.S. blood supply.
Cytomegalovirus.
CMV is carried in and transmitted by lymphocytes. In most patients transfusion-associated CMV infection causes no symptoms, although it may cause a mild mononucleosis-type illness 3 to 4 weeks after transfusion. Transfusion of CMV-containing blood products into an immunocompromised host with no serologic evidence of prior exposure to CMV can cause a lethal systemic infection. Patients at risk for severe CMV infection include bone marrow transplant patients, organ transplant candidates and recipients, and high-risk neonates. Therefore patients who have never been infected with CMV and have an inherited or acquired immunodeficiency state should receive CMV-negative or leukoreduced RBCs.
Babesia.
Babesia spp., especially Babesia microti, is responsible for more transfusion-transmitted infectious diseases than any other pathogen in the United States. Many infections are asymptomatic, but disease can occur, especially in immunocompromised patients such as premature infants and patients without functional spleens. Symptoms generally occur 1 to 9 weeks after infection and can include fever, headache, chills, sweats, myalgia, malaise, and hemolytic anemia; symptoms have progressed to death in at least 12 cases of transfusion-transmitted babesiosis in the USA. Although transfusion-transmitted babesiosis predominantly occurs in the endemic areas of the northeast and upper midwest United States, it is not restricted to those regions because both blood donors and units of blood travel.
Bacteria (Platelet Units).
Platelets are associated with the same range of infectious pathogens as any other blood component, but septic transfusion reactions, caused by bacterially contaminated units, comprise a unique risk of platelet transfusion. In contrast to all other blood components, which are stored either at 4° C or frozen, platelets are stored at room temperature. The reason is that if platelets are chilled before transfusion, they are cleared rapidly from the recipient’s circulation. Although room temperature storage dramatically improves platelet survival in vivo, it also tends to promote bacterial growth. Currently, bacteria can be cultured from approximately 1 in 5000 apheresis platelet units. In the United States blood donor centers generally use culture methods to screen for bacteria in all platelet products, although alternative rapid testing methods are also available. Using current screening methods, the per-unit risk of a clinically apparent septic transfusion reaction is estimated to be 1 in 75,000 platelet transfusions.
Platelet Transfusions
Indications for Transfusion
Bone Marrow Failure
Bone marrow failure resulting in thrombocytopenia is the major reason for patients to receive platelet transfusions. Iatrogenic induction of bone marrow failure, secondary to chemotherapy or irradiation, is the most common cause of bone marrow failure in all age groups. Patients with malignancies involving the bone marrow (e.g., leukemia, lymphoma, neuroblastoma, metastatic disease from solid tumors, myelofibrosis) may also require platelet support. The decreased marrow production in these diseases is caused by displacement of megakaryocytes with malignant elements. In some cases the tumor cells produce substances (e.g., tumor necrosis factor) that inhibit the growth of normal marrow cells. If there is no evidence of platelet dysfunction, patients usually do not experience active bleeding if the platelet count is greater than 10,000/mm 3 and have minimal risk for spontaneous life-threatening hemorrhage until the count is less than or equal to 5000/mm 3 . Although the bleeding rate is higher in patients undergoing pediatric oncology and bone marrow transplant procedures, the bleeding risk is not closely associated with the platelet count, and transfusion triggers of 10,000/mm 3 are still recommended for pediatric patients . However, additional coagulation abnormalities, such as acquired or congenital platelet dysfunction, clotting protein deficiencies, and vascular or tissue defects, require more aggressive platelet support to decrease the risk of hemorrhage.
Patients with primary bone marrow failure caused by a defect such as Fanconi anemia or aplastic anemia will also be thrombocytopenic. However, platelets are usually withheld from these patients unless they are actively bleeding because they have a high rate of alloimmunization to HLA antigens, which in turn will significantly reduce the success of future transfusions and complicate bone marrow transplants. Studies conducted before the introduction of modern leukoreduction filters found that exposure of patients with aplastic anemia to more than five platelet donors results in a significant reduction in successful transplantation owing to rejection of the donor bone marrow. Although it is unclear whether this finding applies to current leukoreduced platelets, if these patients do require treatment for bleeding, it is best to use irradiated, single-donor, leukoreduced platelets in an attempt to minimize sensitization. The decision to transfuse a patient who has been newly diagnosed with aplastic anemia should not be taken lightly because even a single exposure to transfused platelets may adversely affect long-term survival if bone marrow transplantation fails. Once the patient has become refractory to single-donor and HLA-matched platelets, family donors might also undergo plateletpheresis if the patient is not a candidate for bone marrow transplantation.
Platelet Destruction
Shortened platelet survival may result from immune-mediated destruction or nonimmune mechanisms, leading to accelerated platelet consumption ( Table 36-8 ). In general, prophylactic platelet transfusions are contraindicated in these patients because the infused platelets are rapidly destroyed and thus a “safe” platelet count cannot be maintained. Therefore platelet transfusions are reserved for episodes of active bleeding or in preparation for surgical procedures.
| Alloantigen System | Glycoprotein Location | Allelic Forms | Phenotypic Frequency |
|---|---|---|---|
| HPA-1 (P1 A , Zw) | GPIIIa Leu–Pro33 | HPA-1a (P1 A1 )/HPA-1b (P1 A2 ) | 72% a/a |
| 26% a/b | |||
| 2% b/b | |||
| HPA-2 (Ko, Sib) | GP1b Thr–Met145 | HPA-2a (Ko b )/HPA-2b (Ko a ) | 85% a/a |
| 14% a/b | |||
| 1% b/b | |||
| HPA-3 (Bak, Lek) | GPIIb Ile–Ser843 | HPA-3a (Bak a )/HPA-3b(Bak b ) | 37% a/a |
| 48% b/b | |||
| 15% b/b | |||
| HPA-4 (Pen, Yuk) | GPIIIa Arg–Gln143 | HPA-4a (Pen a )/HPA-4b(Pen b ) | 99% a/a |
| <0.1% a/b | |||
| <0.1% b/b | |||
| HPA-5 (Br, Hc, Zav) | GPIa Glu–Lys505 | HPA-5a (Br b )/HPA-5b (Br a ) | 80% a/a |
| 19% a/b | |||
| 1% b/b | |||
| HPA-6 (Ca, Tu) | GPIIIa Arg–Gln489 | HPA-6a (Ca b )/HPA-6b (Ca a ) | 99% a/a |
| <1% a/b | |||
| <1% b/b | |||
| HPA-7 (Mo) | GPIIIa Pro–Ala407 | HPA-7a (Mo b )/HPA-7b (Mo a ) | 99% a/a |
| <1% a/b | |||
| <1% b/b | |||
| HPA-8 (Sr a ) | GPIIIa Arg–Cys636 | HPA-8a (Sr b )/HPA-8b (Sr a ) | 99% a/a |
| <1% a/b | |||
| <1% b/b |
Nonimmune Mechanisms.
Nonimmune mechanisms include the utilization of platelets in areas of endothelial damage, such as hemolytic-uremic syndrome (HUS), thrombotic thrombocytopenic purpura (TTP), or NEC. DIC results in decreased platelet life span owing to consumption of platelets and activated clotting factors, such as fibrinogen in the formation of microthrombi in association with sepsis, shock, acidosis, or severe head trauma. In these syndromes it is essential to assess the risk of bleeding in determining the need for transfusion. Infants with NEC are often transfused more aggressively because many demonstrate marked gastrointestinal blood loss and may have a preexisting central nervous system hemorrhage associated with prematurity. Similarly, patients with DIC are often given platelets even though their counts may not be less than 10,000/mm 3 , owing to consumption of clotting factors and evidence of ongoing bleeding. Successful outcome for these patients depends on elimination of the underlying disease—for example, treating sepsis with antibiotics, restoration of organ perfusion in states of shock, or removal of necrotic tissue. Patients with large cavernous sinus hemangiomas (Kasabach-Merritt syndrome) have localized consumption of platelets and clotting factors, resulting in a laboratory picture of DIC. Until the lesion is removed or begins to involute, the platelet and clotting factor consumption will continue. These patients usually do not require transfusion for thrombocytopenia because the high rate of platelet turnover results in younger and more hemostatically effective platelets in circulation. Finally, in TTP and HUS, platelet infusions are withheld as long as possible because it is thought that the infused platelets would lead to further hemolysis and organ damage by contributing to the formation of microthrombi in areas of endothelial damage.
Hypersplenism may result in poor platelet survival owing to trapping of platelets in the expanded splenic bed. A more complex clinical situation arises with acute or chronic liver disease. Splenic pooling, DIC, and poor platelet production may all contribute to the thrombocytopenia of hepatic disease. The presence of portal hypertension and esophageal varices from cirrhosis may be an indication to consider platelet transfusion in the presence of gastrointestinal bleeding or when the platelet count falls below 20,000/mm 3 . Acquired platelet dysfunction is associated with both acute and chronic liver disease owing to decreased hepatic clearance of circulating fibrin degradation products. When platelet transfusion is required, splenomegaly may affect initial platelet recovery measured in the first hour after transfusion, but not platelet survival as determined by the platelet count 24 hours later.
Immune-Mediated Thrombocytopenia.
In immune thrombocytopenia antibodies bind to epitopes on platelet membranes resulting in rapid removal of the platelets from the circulation. These antibodies are either “self-reactive” autoantibodies or specific for alloantigens, polymorphic epitopes expressed on platelet membrane receptors. Alloantibodies cause thrombocytopenia in neonatal alloimmune thrombocytopenia and posttransfusion purpura (PTP), both of which are discussed later in this chapter. Alloantibodies responsible for poor response to platelet transfusion are discussed in the section on platelet refractory state.
Autoantibody-Mediated Thrombocytopenia.
Autoantibody to platelets may be present in association with other diseases, such as systemic lupus erythematosus, Epstein-Barr virus infection, CMV infection, varicella virus infection, multiple sclerosis, diabetes, thyroid disease, and drug-induced autoantibody as seen with valproic acid. It may also be present without other disease associations, as in idiopathic thrombocytopenic purpura. In either setting there is rapid removal of antibody-coated platelets in the spleen and, to a lesser extent, in the liver. Immune complexes may nonspecifically adhere or bind by means of Fc receptors on the platelet surface. A particularly interesting example of the latter is the entity heparin-induced thrombocytopenia (HIT). In this case patients exposed to heparin therapy produce antibodies directed to platelet factor 4 (PF4) bound to heparin. Immune complexes of PF4-heparin and antibody bind via Fc receptors to platelets, causing platelet activation and a prothrombotic state in which platelets are consumed. This may be a devastating complication of heparin therapy that may be suspected in patients who show falling platelet counts a few days after initiating heparin therapy. Platelet transfusions are generally contraindicated insofar as they may exacerbate the prothrombotic state.
In states that increase platelet turnover, the bone marrow responds by increasing megakaryocyte number, size, and ploidy. This, in turn, results in fresher platelets that are slightly larger in size and represent a younger population than normal owing to the rapid turnover of thrombocytes. Thus the bleeding time in patients with immune-mediated thrombocytopenia is shortened, relative to the absolute number of platelets, and such patients rarely experience major bleeding even though the platelet count may fall below 10,000/mm 3 . Autoantibodies that interfere with functional epitopes on platelet glycoprotein receptors, such as in acquired Glanzmann thrombasthenia, are a notable exception to this rule. In this setting patients present with bleeding even with platelet counts greater than 20,000/mm 3 .
In patients with rapid consumption of platelets by autoantibody, platelet transfusions are generally not indicated unless there is evidence of bleeding, regardless of the platelet count. Central nervous system and gastrointestinal bleeding may be the presenting features of autoantibody-mediated thrombocytopenia and require immediate aggressive therapy, including platelet transfusion. Although the platelets are rapidly consumed, active bleeding may stop or decrease significantly. Platelet transfusions should be used judiciously in combination with therapy aimed at reducing autoantibody production and blocking reticuloendothelial cell clearance of antibody-coated cells. The therapy for immune-mediated thrombocytopenia is discussed elsewhere.
Alloantibody-Mediated Thrombocytopenia—Neonatal Alloimmune Thrombocytopenic Purpura.
In neonatal alloimmune thrombocytopenic purpura (NATP), maternal antiplatelet alloantibody has crossed the placenta, resulting in thrombocytopenia in the fetus and neonate. Harrington and Sprague first reported the observation that pregnant women may become alloimmunized to antigens expressed on fetal platelets. Even during the first pregnancy, maternal antiplatelet alloantibodies can cross the placenta and cause profound thrombocytopenia in the fetus and newborn. In North America the alloantibodies identified are most commonly directed against human platelet antigen 1a (HPA-1a, also known as PLA-1). A complete list of platelet alloantigens and their frequency in those populations that have been studied thus far are listed in Table 36-8 . The presentation of thrombocytopenia in a healthy infant whose mother has a normal platelet count is often the result of maternal alloantibodies.
The diagnosis of NATP is made with the demonstration of antiplatelet antibody in maternal plasma, reactive with paternal platelets. Maternal plasma is also screened to rule out the presence of autoantibody. More than 50% of the platelet-specific alloantibodies in NATP are reactive with the HPA-1a epitope on glycoprotein IIIa. Healthy infants may not bleed or develop petechiae until the platelet count drops below 20,000/mm 3 . Reports suggest that immune response genes may also play a role in platelet alloimmunization. On the basis of these studies, there appears to be an increased incidence of alloimmunization against HPA-1a in mothers positive for HLA-B8 and HLA-DR3.
The diagnosis of maternal alloimmunization is usually made after the birth of a mother’s first thrombocytopenic infant. Women who are then identified as sensitized to a platelet-specific alloantigen should be monitored very closely throughout subsequent pregnancies. Intermittent screening of maternal serum should be performed similar to prenatal Coombs testing in women who are Rh negative. All women in the mother’s family should also be evaluated before or during their pregnancies to determine whether they might also be at risk of alloimmunization. If a HPA-1a–negative woman is also HLA-DR3 (DRw52) positive, she has a greatly increased risk of alloimmunization even during her first pregnancy.
To determine whether the infant is at risk for thrombocytopenia secondary to maternal alloantibody, it is advantageous to establish the platelet phenotype of the fetus early in pregnancy. As early as the eighth week of gestation, DNA from nucleated cells obtained from chorionic villus sampling can be amplified through the use of the polymerase chain reaction. DNA typing is then determined with glycoprotein-specific oligonucleotide primers and probes unique for each alloantigen sequence or phenotype. Results from a prospective study described a 100% correlation between DNA typing and subsequent serologic response in mothers with alloantibodies to PlA1 or Bak. This technique is useful only where the nucleotide sequences that define the polymorphisms are known (see Table 36-8 ).
Because of the high frequency of these alloantigens, random donor platelets are usually ineffective in treating alloimmune thrombocytopenia in the neonate. When platelets are given, it is best to use platelets lacking the implicated antigen. If the antigen is unknown or antigen-negative platelets are unavailable, then washed, irradiated maternal apheresis platelets can be used because they lack the alloantigen. Washing is required to remove maternal plasma that contains the alloantibody. Irradiation is routinely performed on all blood products from first-degree relatives where there is an increased risk of GVHD, particularly in neonates. When severe thrombocytopenia in the newborn is anticipated, antigen-negative platelets may be procured before elective delivery and made available for transfusion shortly after birth for infants with NATP diagnosed prenatally. Intravenous immunoglobulin has also been used successfully in the newborn. In families with no history of prenatal intracranial hemorrhage, fetal platelet count should be closely monitored and ultrasonography performed to rule out bleeding before delivery.
Platelet Transfusions for Surgery
Retrospective data suggest that a platelet count above 50,000/mm 3 is acceptable for a patient to undergo major surgery. Certain circumstances may warrant more aggressive correction of thrombocytopenia before or after surgery. In areas where even a small amount of bleeding might cause permanent deficits, such as the eye or central nervous system, it is traditional to aim for a platelet count higher than 100,000/mm 3 . During prolonged procedures where the patient will be on cardiopulmonary bypass or extracorporeal membrane oxygenation (ECMO), platelets may be dysfunctional owing to activation after passage through extracorporeal perfusion equipment. In general, there is no need to administer platelets to patients after extracorporeal circulation if there is no evidence of bleeding and the platelet count is at least 50,000/mm 3 . When bleeding is present, it is more commonly caused by heparin, inadequate protamine correction, fibrinolysis, or problems with surgical closure.
Minor procedures such as liver biopsies have been performed safely with platelet counts greater than 25,000/mm 3 . Similarly, lumbar puncture has been shown to be safe in the setting of thrombocytopenia. Howard and colleagues reported a series of 956 consecutive pediatric patients with newly diagnosed acute lymphoblastic leukemia who underwent lumbar puncture. No serious bleeding complications occurred in 5223 lumbar punctures, including 170 procedures performed when the platelet count was 11,000 to 20,000/µL. The authors concluded that prophylactic platelet transfusion was unnecessary in patients with platelet counts above 10,000/µL. These procedures were performed in only 29 patients with platelet counts below 10,000/µL, so it was not possible to assess with confidence the risk of bleeding in patients with extremely low platelet counts.
Platelet Transfusions for Patients with Dysfunctional Platelets
Patients with congenital platelet dysfunction, such as Glanzmann thrombocytopathy or Bernard-Soulier syndrome, may experience episodes of profuse bleeding. In this setting platelets may be administered to control massive hemorrhage. Because normal platelets express proteins that these individuals lack, some patients develop isoantibodies to these receptors and eventually become refractory to all platelet transfusions. Efforts to minimize menstrual bleeding using hormonal suppression may decrease the number of platelet infusions in women. Bleeding in patients with congenital platelet dysfunction caused by abnormal platelet secretion may respond to desmopressin, avoiding exposure to blood.
Acquired platelet dysfunction in children is most commonly caused by drugs, chronic renal failure, or antiplatelet antibodies. Rarely, children present with platelet dysfunction as a manifestation of myelodysplastic syndrome or chronic leukemia. Platelet transfusions may be used if the patient is actively bleeding, but eventually the transfused platelets will be affected by exposure to drug, uremia, or antiplatelet antibodies, which limit their survival and function. For this reason platelets are not given prophylactically in these patients. Every effort should be made to ameliorate the platelet dysfunction so that exposure to blood product is avoided. Patients with uremia may respond to infusions of desmopressin, and those with antiplatelet autoantibodies may respond to immunoglobulin infusions or corticosteroid therapy. When possible, alternative medications should replace those known to cause platelet dysfunction.
Platelet Units
Platelets are prepared for transfusion from a single unit of freshly collected whole blood (whole blood–derived [WBD] platelets) or from apheresis by means of continuous-flow centrifugation (apheresis platelets). Regardless of the technique used, platelets must always be collected and stored at 20 to 24° C and in anticoagulant-containing citrate-phosphate-dextrose (CPD) or CPDA-1 or an additive solution that may contain some combination of acetate, magnesium, potassium, or gluconate. Citrate anticoagulates the blood by binding plasma calcium, whereas dextrose is a nutrient source for actively metabolizing platelets.
Whole Blood–Derived Platelets
In the United States WBD platelets are prepared from whole blood by the platelet-rich plasma (PRP) method. PRP is separated from packed RBCs by centrifugation of whole blood (“soft spin”). The PRP is then centrifuged again (“hard spin”). All but 50 to 70 mL of the platelet-poor plasma (PPP) is transferred to an adjoining bag to be used for the production of fresh frozen plasma or other blood components, such as cryoprecipitate or albumin. When optimal centrifugation techniques are used, approximately 85% of the platelets (5 to 7 × 10 10 platelets) are recovered from a unit of whole blood. In Canada and Europe the alternate “buffy coat” method is used to generate platelet concentrates from whole blood.
Apheresis Platelets
Apheresis platelets from one individual should yield more than 3 × 10 11 platelets in 200 to 300 mL of plasma. Modern apheresis equipment is often able to yield more than 6.5 × 10 11 platelets per procedure and typically incorporates a leukoreduction step that leaves fewer than 1 × 10 6 leukocytes per product. This allows units to be split into two or even three apheresis platelet units, each with five to six WBD platelet equivalents.
Once they are processed, platelet units are stored on elliptical, circular, or flat-bed agitators. This constant mild agitation prevents platelet clumping or activation as platelets settle to the bottom of the bag. Constant agitation also facilitates gas exchange, which is important for maintaining an optimal pH range for platelet metabolism and function at 20°C. Because of the risk of bacterial growth associated with room temperature storage, platelet units are usually stored for no more than 5 days.
Platelet Dosing
Guidelines for determining platelet dose have been published in a number of reviews. In general, one unit of random donor platelets (5 to 7 × 10 10 platelets) per 12 kg of body weight should increase the platelet count by 40,000 to 50,000/mm 3 within 1 hour after the infusion. In the average-size adolescent or adult, 6 units of platelet concentrate, or 1 single-donor apheresis unit (3 to 5 × 10 11 ), will increase the platelet count by 50,000/mm 3 or greater. This dose or a lower dose are equivalent in bleeding outcomes, whereas a higher dose increases the number of platelets transfused without any benefit in bleeding outcomes. Concentrating or washing the platelets can result in variable platelet loss and decrease the platelet count increments after transfusion.
To determine the effectiveness of platelet transfusion, a platelet count should be obtained before infusion, at 1 hour, and at 24 hours after infusion. There is no significant difference in the posttransfusion platelet count when measured at 10 minutes versus 60 minutes after transfusion.
Platelet Refractory State
When a patient no longer achieves the expected rise in platelet count after a platelet transfusion, the clinician must first assess whether the shortened platelet recovery and survival result from immune or nonimmune mechanisms. Examples of nonimmune mechanisms are listed in Table 36-8 and are responsible for the majority of cases in which posttransfusion count increments are lower than expected. The most notable of these include splenomegaly, DIC, and fever.
Normally, one third of the platelets released into circulation from the bone marrow are stored in the spleen. Hypersplenism causes a larger percentage of transfused platelets to be sequestered in the spleen. Hypersplenism results in a decrease in the platelet recovery immediately after platelet infusion but does not have an effect on platelet survival once a steady state has been achieved.
Fever may reduce platelet recovery by 20% to 40%. In one study decreased platelet recovery was documented in patients who underwent bone marrow transplant with fever and negative blood cultures. Although occult infection cannot be ruled out, in these patients fever alone was the only change associated with poor platelet increment.
Consumptive coagulopathy (DIC) refers to the excessive activation of thrombin resulting in an accelerated conversion of fibrinogen to fibrin. This process may be acute and accompanied by rapid consumption of procoagulant factors and platelets in association with extensive bleeding, as in sepsis, shock, or certain forms of malignancy. A chronic form of DIC may occur if the procoagulant consumption is slow enough for the liver to compensate for a limited consumption, as found in hemangiomas and necrotic lesion in the lung or brain, for example. In either form of DIC, recovery and survival of transfused platelets is shortened. Diseases associated with primary endothelial cell damage, such as HUS and TTP, are also associated with a decreased platelet recovery owing to the formation of microaggregates in the areas of vascular damage. Additional bleeding may be seen with these syndromes because of the acquired platelet dysfunction associated with renal failure and uremia.
Older platelets may not remain in circulation as long as freshly collected platelet concentrates because of excessive activation during storage or metabolic effects of acidosis. Platelets also express small amounts of ABO antigens, making ABO-incompatible transfusions slightly less effective than ABO-matched transfusions. Finally, analysis of multiply transfused patients show that there is a tendency to decreased average count increments with successive transfusions. A patient’s first platelet transfusion may result in a 30% higher count increment than their fifteenth, despite equal numbers of platelets being transfused and in the absence of other factors. No definitive cause of this decrease has been discerned, other than the hypothesis that patients subjected to multiple rounds of chemotherapy may sustain subtle endothelial damage that consumes transfused platelets.
To compare the effectiveness of platelet transfusion on different occasions or with different donors and recipients, the best ratio to use is the corrected count increment (CCI):
CCI = ( Posttransfusion count − Pretransfusion count ) ( body surface area m 2 ) Number of platelets given ( × 10 11 )
Related posts:
 The Neonatal Erythrocyte and Its Disorders
The Neonatal Erythrocyte and Its Disorders
 Diagnostic Approach to the Anemic Patient
Diagnostic Approach to the Anemic Patient
 Approach to the Child with a Suspected Bleeding Disorder
Approach to the Child with a Suspected Bleeding Disorder
 Disorders of the Red Cell Membrane
Disorders of the Red Cell Membrane
 Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
 Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


