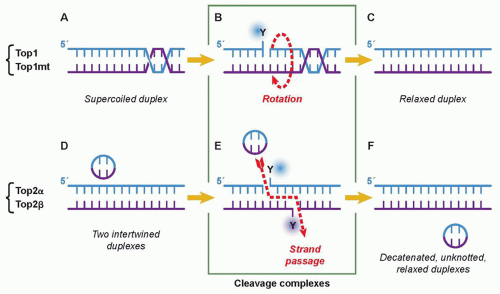
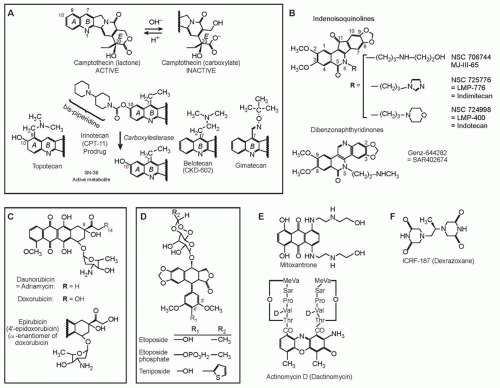
Cytotoxic Mechanisms of Top2cc-Targeted Drugs (Intercalators and Demethyl Epipodophyllotoxins)
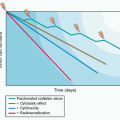
Contrary to camptothecins, Top2 inhibitors kill cancer cells without requiring DNA replication fork collisions. Indeed, even after a 30-minute exposure, doxorubicin and other Top2cc-targeted drugs can kill over 99% of the cells, which is in vast excess of the fraction of S-phase cells in tissue culture (generally less than 50%).
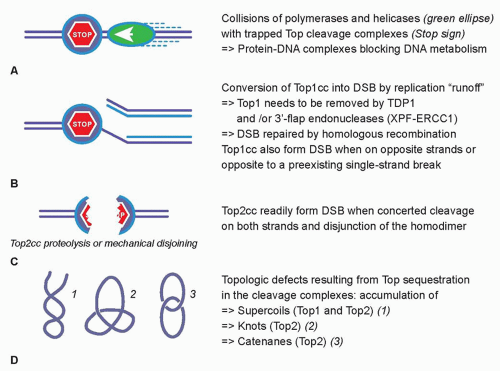
27,28 The collision mechanism in the case of Top2cc-targeted drugs (see
Fig. 20.2A) appears to involve transcription and proteolysis of both Top2 and RNA polymerase II.
29 Such situation would then lead to DNA double-strand breaks by disruption of the Top2 dimer interface (see
Fig. 20.2C). Alternatively, the Top2 homodimer interface could be disjoined by mechanical tension (see
Fig. 20.2C). Yet, it is important to bear in mind that 90% of Top2cc trapped by etoposide are not concerted and, therefore, consist in single-strand breaks,
3,30,31 which is different from doxorubicin, which traps both Top2 monomers and produces a majority of DNA double-strand breaks.
32 Finally, it is not excluded that topologic defects resulting from Top2 sequestration by the drug-induced cleavage complexes could contribute to the cytotoxicity of Top2cc-targeted drugs (see
Fig. 20.2D). Such topologic defects would include persistent DNA knots and catenanes, potentially leading to chromosome breaks during mitosis.