The Skeletal System in Thyrotoxicosis
Graham R. Williams
Regulation of Thyroid Status
Circulating thyroid hormone concentrations are maintained in the euthyroid range by the hypothalamic–pituitary–thyroid (HPT) axis, a classic example of an endocrine negative feedback loop (Fig. 28.1). Thyrotropin-releasing hormone (TRH) is synthesized and released from neurons in the paraventricular nucleus of the hypothalamus. TRH passes into the portal system and travels to the anterior pituitary where it acts on TRH receptors in thyrotropes to stimulate production and secretion of thyrotropin [thyroid-stimulating hormone (TSH)]. TSH then acts on the G-protein–coupled TSH receptor in thyroid follicular cells to stimulate their growth and the synthesis and release of the thyroid hormones, thyroxine (T4), and 3,5,3′-L-triiodothyronine (T3) (1). Thyroid hormones inhibit synthesis and secretion of TRH and TSH in the hypothalamus and pituitary, thus completing the negative feedback loop and maintaining circulating thyroid hormones and TSH in a physiologic inverse relationship that defines the set-point of the HPT axis (2).
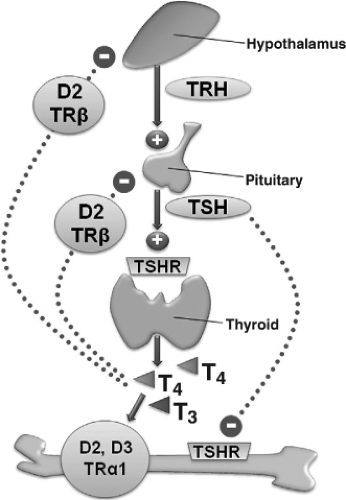 Figure 28.1 Relationship between the hypothalamic–pituitary–thyroid axis and bone. The actions of thyroid hormone are mediated by TRα1 in bone but by TRβ in the hypothalamus and pituitary. The actions of TSH are mediated by the TSH receptor (TSHR), which is expressed in thyroid follicular and bone cells. Serum concentrations of thyroid hormones and TSH are maintained in an inverse relationship by the HPT axis feedback loop. T3 stimulates bone cell activities, whereas TSH is proposed to inhibit bone cell function. The type 2 deiodinase enzyme (D2) converts inactive T4 to active T3 in T3 target cells in bone, hypothalamus, and pituitary and plays a critical role in controlling intra-cellular availability of T3. The type 3 deiodinase (D3) inactivates T3 in target cells in bone acting as an additional regulator of T3 action in the skeleton. “Reprinted from, Archives of Biochemistry and Biophysics, Vol 503, Gogakos AI, Bassett JHD, Williams GR, Thyroid and bone, 129–136, Copyright 2010, with permission from Elsevier.” |
Normal individuals each have a unique HPT axis set-point that lies within the population reference range. The range of thyroid status in an individual over time varies by less than 50% of the reference range for the population as a whole (3), indicating that there are differences between normal individuals in the sensitivities of specific tissues to thyroid hormones (4). Heritability of the thyroid status in twin studies is estimated at 23% to 64% for circulating free T3 concentrations, 39% to 65% for free T4 levels, and 64% to 65% for TSH (5,6). A genome wide screen identified eight quantitative trait loci linked to FT3, FT4, and TSH levels, and a recent association study of 68 candidate thyroid pathway genes revealed several associations with TSH and free T4 levels, indicating that thyroid status is inherited as a complex genetic trait (7,8). Similar genetic heterogeneity is also likely to regulate and fine-tune tissue-specific thyroid hormone sensitivity, including bone.
Control of Intracellular T3 Availability
The thyroid gland mainly secretes the pro-hormone T4 but also produces smaller amounts of the active hormone T3. Most of the circulating T3 is generated via 5′-deiodination of T4, a reaction catalyzed by the type 1 iodothyronine deiodinase enzyme (Dio1) in liver and kidney. Over 95% of thyroid hormones are bound to plasma proteins and the circulating free T4 concentration is maintained at approximately 3- to 4-fold higher level than the concentration of free T3. Entry of T3 and T4 into target cells is determined by active uptake of free hormones mediated by specific cell membrane transporters including monocarboxylate transporter-8 (MCT8), MCT10, and organic acid transporter protein-1c1 (OATP1c1) (9). The intracellular availability of the active hormone T3 to nuclear thyroid hormone receptors (TRs) is determined by the relative activities of the type 2 and 3 deiodinase enzymes (Dio2, Dio3). Dio2 converts
T4 to T3 by catalyzing removal of a 5′-iodine atom, whereas Dio3 prevents activation of T4 and inactivates T3 by removal of a 5-iodine atom to generate the metabolites 3,3′,5′-L-triiodothyronine (reverse T3) and 3,3′-diiodothyronine (T2), respectively (10). Thus, pre-receptor control of ligand (T3) availability to the TR in target cells is a key mechanism that controls the magnitude of cellular responses to thyroid hormones in a tissue-specific manner.
T4 to T3 by catalyzing removal of a 5′-iodine atom, whereas Dio3 prevents activation of T4 and inactivates T3 by removal of a 5-iodine atom to generate the metabolites 3,3′,5′-L-triiodothyronine (reverse T3) and 3,3′-diiodothyronine (T2), respectively (10). Thus, pre-receptor control of ligand (T3) availability to the TR in target cells is a key mechanism that controls the magnitude of cellular responses to thyroid hormones in a tissue-specific manner.
Mechanism of Thyroid Hormone Action
The major actions of thyroid hormone are mediated by TRα1, TRβ1, and TRβ2 nuclear receptors, encoded by the THRA and THRB genes, which act as hormone inducible transcription factors that regulate the expression of T3-responsive target genes (11). In the absence of hormone, unliganded TRs mediate transcriptional repression of target genes, and binding of T3 to the receptor results in de-repression and activation of gene transcription. TRα1 and TRβ1 are expressed in most tissues but their relative concentrations differ between cell types and at different times during development and adulthood (12). By contrast, TRβ2 expression is restricted to the hypothalamus and pituitary where it mediates negative feedback inhibition of TRH and TSH, playing a key role in regulation of the HPT axis (13,14).
TRα1 and TRβ1 are both expressed in bone and cartilage but TRα1 predominates and is expressed at concentrations that are at least 10-fold higher than TRβ1 (15,16). Both TR isoforms are present in growth plate chondrocytes and bone-forming osteoblasts, but it is currently unclear whether osteoclasts also express TRs and respond to T3 (17,18). Studies in mice with mutation or deletion of the THRA or THRB genes encoding TRα or TRβ have demonstrated that TRα is the principal mediator of T3 action in bone (16,18,19,20,21,22,23).
Bone Remodeling Cycle
The structure and strength of the skeleton is maintained by a continuous process of bone remodeling, which is achieved by the integrated activities of the three principal cell types in the skeleton; osteocytes, osteoclasts, and osteoblasts (Fig. 28.2). Osteocytes are derived from osteoblasts that have become embedded in bone matrix and comprise over 90% of all adult bone cells. The osteocyte network senses changes in mechanical load and regulates local initiation of bone remodeling via the release of cytokines and chemotactic signals. Bone remodeling commences with recruitment of mature and precursor osteoclasts to sites of mechanical strain or micro-damage. Osteoclasts excavate a resorption cavity over a period of 3 to 5 weeks until they eventually undergo apoptosis to terminate the process and signal the recruitment of osteoblast precursors. Osteoblasts migrate to excavation sites, where they mature and secrete and mineralize osteoid to replace the resorbed bone over a period of bone formation that lasts about 3 months. Coupling of osteoclast and osteoblast activities via signaling between the two cell lineages regulates this cycle of bone remodeling and results in skeletal homeostasis with preservation of bone strength. In summary, the bone remodeling cycle is initiated and orchestrated by osteocytes, and regulated by coupled crosstalk between osteoblasts and osteoclasts.
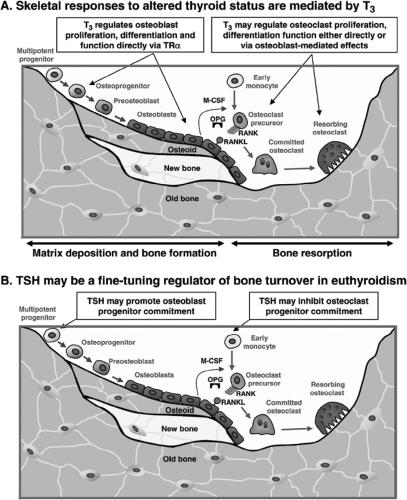 Figure 28.2 Proposed regulatory roles for T3 (panel A) and TSH (panel B) in bone remodeling. The bone remodeling cycle is initiated and orchestrated by osteocytes which are embedded within mineralized bone and communicate via ramifications of dendritic processes. Coupled crosstalk between bone-forming osteoblasts and bone resorbing osteoclasts regulates bone remodeling to maintain skeletal homeostasis. M-CSF, macrophage colony stimulating factor; OPG, osteoprotegerin; RANK, receptor activator of NFκB; RANKL, RANK ligand. “Reprinted from, Bone, Vol 43, Bassett JHD, Williams GR, Critical role of the hypothalamic–pituitary–thyroid axis in bone, 418–426, Copyright 2008, with permission from Elsevier.” |
Thyrotoxicosis results in a state of high bone turnover with increased bone resorption and formation rates. The frequency of initiation of bone remodeling is markedly increased and the duration of the bone remodeling cycle is reduced. The net result is that the duration of bone formation and mineralization is reduced to a greater extent than the reduction in duration of bone resorption. This leads to a net 10% loss of bone per remodeling cycle, resulting in high bone turnover osteoporosis (24). Accordingly, biochemical markers of bone formation [serum alkaline phosphatase, osteocalcin, and carboxyterminal propeptide of type 1 collagen (P1NP)] and resorption [urinary pyridinoline and deoxypyridinoline collagen cross-links, hydroxyproline and carboxyterminal cross-linked telopeptide of type 1 collagen (CTX)] are all elevated in hyperthyroidism and generally correlate with disease severity (25,26,27,28,29).
These detailed analyses of bone turnover in thyroid disease were performed many years ago, but understanding of the molecular mechanisms that mediate thyroid hormone regulation of bone remodeling remains limited. Although it is well known that thyrotoxicosis results in increased osteoclastic bone resorption (30), it has not been determined whether T3 acts directly in osteoclasts (31,32). Thus, it is possible that effects of thyroid hormones on osteoclastic bone resorption are indirect and mediated primarily by direct actions of T3 in osteoblasts or other target cells in the bone marrow microenvironment (Fig. 28.2A). In osteoblasts, T3 regulates cell differentiation (33,34) and bone matrix synthesis and degradation (35), thereby acting as an essential regulator of bone mineralization and strength (36).
The actions of thyroid hormones on bone and their effects on calcium homeostasis also result in a negative calcium balance in thyrotoxicosis (37). Urinary calcium excretion and phosphate reabsorption are increased along with reduced intestinal calcium and phosphate absorption and elevated fecal calcium loss. Serum albumin concentrations are reduced, resulting in elevated mean ionized calcium and phosphate concentrations. Nevertheless, marked hypercalcemia is uncommon, although mild hypercalcemia may be present in up to 20% of patients. This relative hypercalcemia suppresses parathyroid hormone (PTH) secretion and serum PTH concentrations correlate negatively with FT4 levels. There is also reduced renal conversion of 25(OH)-vitamin D3 to 1α,25(OH)2-vitamin D3, as the reaction requires PTH. 1α,25(OH)2-vitamin D3 levels are decreased further by increased metabolic clearance in thyrotoxicosis. In combination with the calcium-mobilizing effects of thyroid hormones on bone, these changes all contribute to significant negative calcium balance in hyperthyroidism (29,37).
TSH and Bone
Although the TSHR is primarily expressed in thyroid follicular cells, its presence has also been reported in other tissues including brain, kidney, testis, heart, adipose tissue, fibroblasts, hemopoietic cells, immune cells, and bone (38). In the skeleton, TSHR expression has been demonstrated in both osteoblasts and osteoclasts (39), suggesting TSH may exert direct actions in bone (Fig. 28.2B). Indeed, TSH was proposed as a negative regulator of bone remodeling that acts directly in skeletal cells to inhibit bone turnover (39). Knockout mice
lacking TSHR displayed a phenotype of high bone turnover osteoporosis, and in cell culture studies TSH inhibited activities of both osteoblasts and osteoclasts via cyclic adenosine monophosphate (cAMP)-independent pathways. These experiments led to the proposal that bone loss in thyrotoxicosis results from absent or reduced inhibitory actions of TSH on the skeleton in contrast to the consequences of thyroid hormone excess (39). However, additional studies have shown TSH either stimulated (40,41) or had no effect (42) on osteoblast differentiation, whilst effects on osteoclasts were either inhibitory (40,41,43) or absent (42). Analysis of the TSHR expressed in osteoblasts and osteoclasts subsequently revealed that receptor protein expression was very low compared to levels in thyroid follicular cells and that TSH treatment did not stimulate cAMP activity (42,43,44). Nevertheless, TSH treatment does appear to influence tumor necrosis factor-α (TNF-α), activator protein-1 (AP-1), and nuclear factor kappa-light-chain enhancer of activated B cells (NFκB) signaling in bone, suggesting that the TSHR could be coupled to a G-protein other than Gsα in skeletal cells or that TSH might interact with various cytokines to regulate osteoclast and osteoblast activity indirectly.
lacking TSHR displayed a phenotype of high bone turnover osteoporosis, and in cell culture studies TSH inhibited activities of both osteoblasts and osteoclasts via cyclic adenosine monophosphate (cAMP)-independent pathways. These experiments led to the proposal that bone loss in thyrotoxicosis results from absent or reduced inhibitory actions of TSH on the skeleton in contrast to the consequences of thyroid hormone excess (39). However, additional studies have shown TSH either stimulated (40,41) or had no effect (42) on osteoblast differentiation, whilst effects on osteoclasts were either inhibitory (40,41,43) or absent (42). Analysis of the TSHR expressed in osteoblasts and osteoclasts subsequently revealed that receptor protein expression was very low compared to levels in thyroid follicular cells and that TSH treatment did not stimulate cAMP activity (42,43,44). Nevertheless, TSH treatment does appear to influence tumor necrosis factor-α (TNF-α), activator protein-1 (AP-1), and nuclear factor kappa-light-chain enhancer of activated B cells (NFκB) signaling in bone, suggesting that the TSHR could be coupled to a G-protein other than Gsα in skeletal cells or that TSH might interact with various cytokines to regulate osteoclast and osteoblast activity indirectly.
In vivo studies in rodents demonstrated anti-resorptive and anabolic responses to intermittent low doses of TSH, which surprisingly did not affect circulating thyroid hormone concentrations, yet still prevented ovariectomy-induced bone loss (40,41). The possible role of TSH in bone turnover has also been investigated in clinical studies. In patients receiving TSH-suppressive doses of T4 in the management of differentiated thyroid cancer, administration of recombinant human TSH (rhTSH) increases TSH levels to >100 mU/L but does not affect circulating T4 and T3 concentrations as patients have previously undergone total thyroidectomy. In this context, treatment of pre-menopausal women with rhTSH had no effect on serum markers of bone turnover (45,46,47), whilst in post-menopausal women two studies reported a reduction in bone resorption markers accompanied by an increase in bone formation markers following rhTSH administration (46,47), whereas two other studies reported no effect (45,48). Numerous other clinical studies investigating osteoporosis and fracture in relation to altered thyroid status have also been conflicting regarding whether bone loss in thyrotoxicosis results from thyroid hormone excess or TSH deficiency (49). In individuals and animal models in which the HPT axis is intact, and maintains a reciprocal physiologic relationship between thyroid hormones and TSH (Fig. 28.1), this continues to be a difficult question to resolve and the subject is controversial (2). Interpretations of clinical studies have tended to focus either on TSH deficiency or thyroid hormone excess as the underlying cause of bone loss in hyperthyroidism, but either standpoint fails to account for the inverse relationship between thyroid hormones and TSH maintained by the HPT axis (2). A recent large prospective European population study of healthy euthyroid post-menopausal women showed that bone mineral density (BMD) and fracture susceptibility are related to variations in normal thyroid status (50). Thus, thyroid status at the upper end of the normal range in healthy post-menopausal women was associated with lower BMD and an increased risk of non-vertebral fracture. Importantly, even though correlations between BMD and fracture were only evident in relation to changes in thyroid hormone rather than TSH levels, these findings are best interpreted to reflect thyroid status as a whole instead of focusing on the relative importance of thyroid hormone or TSH action in bone (50). A recent study of healthy men aged between 25 and 45 years around the age of peak bone mass similarly demonstrated that higher levels of T3 and T4 within the euthyroid reference range are associated with reduced bone mass and density (51), demonstrating the importance of euthyroid status for bone mineral accrual as well as for bone maintenance (50).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


