Fig. 7.1
The JAK-STAT signalling pathway. Upon binding to the membrane receptors, cytokines initiate downstream signalling cascades through the JAK-STAT pathway. Cytokine receptor homodimers or heterodimers phosphorylate and activate JAKs, which phosphorylate cytoplasmic STAT monomers. Upon phosphorylation, activated STAT dimers translocate to the nucleus and regulate the transcription of target genes
The JAK-STAT pathway is a key intracellular mediator of cytokines that regulate the immune landscape [154]. Dysregulated JAK-STAT signalling, resulting in aberrant expression of STATs and genes regulated by STATs, is a hallmark of many GI cancers. JAK-STAT pathway activity in GI cancers is associated with oncogenesis, proliferation, survival, metastasis, angiogenesis, immune evasion [25, 33, 127], micro-RNA expression [76], and inflammation, which collectively help cultivate an immunosuppressive and tumour-enhancing environment [25]. Thanks to a well-defined ATP-binding pocket that is distinct from other kinases, JAK enzymes are attractive targets for designing highly selective small-molecule inhibitors to modulate STAT-driven molecular effects. A better understanding of JAK-STAT signalling in GI cancers will permit the development of pharmacologic agents with selective profiles that reduce tumour-related inflammation and improve antitumour immunity, as well as inhibit STAT-driven direct oncogenic effects. The objective of this section is to provide an overview of the JAK-STAT components with a focus on evidence concerning the potential role of this pathway in the pathogenesis of GI cancers.
JAK and STAT Family Members
JAKs were named after the Roman god Janus because they are “2-faced,” having a kinase at the carboxyl terminus (at the JH1 domain) and a pseudokinase immediately adjacent at the JH2 domain; a total of 7 homology domains have been identified (JH1–JH7) [1]. The JAK family comprises the following four nonreceptor tyrosine kinases [132]: JAK1 [186], JAK2 [63, 186], JAK3 [82, 87], and tyrosine kinase 2 (TYK2) [42]. JAK1, JAK2, and TYK2 are ubiquitously expressed, whereas JAK3 is restricted primarily to hematopoietic cells [63, 192].
The STAT family comprises the following 7 transcription factors: STAT1–4, STAT5a, STAT5b, and STAT6 [132]. STAT proteins have a conserved tyrosine residue in the carboxy-terminus transactivation domain that is phosphorylated by JAKs and forms STAT dimers by interacting with the SH2 domain; STAT proteins also have coiled coil and DNA binding domains [1]. STATs are ubiquitously expressed, with the exception of STAT4, which is expressed primarily in the thymus and testes [66].
More than 50 cytokines and growth factors signal through various permutations of the 4 JAKs and 7 STATs, leading to a large number of potential signalling outcomes [174]. The downstream signalling effects of key cytokines and growth factors of interest in GI malignancies (Table 7.1; Fig. 7.2) and their potential influence on the tumour microenvironment are explored in a later section of the chapter.
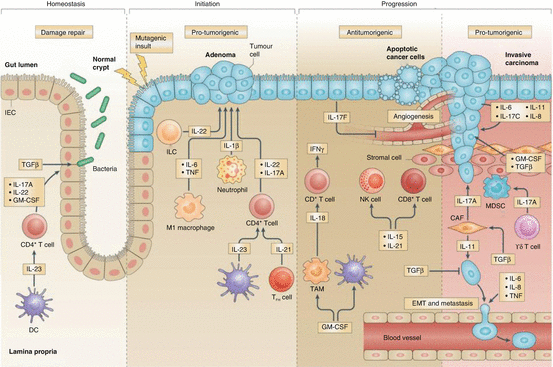
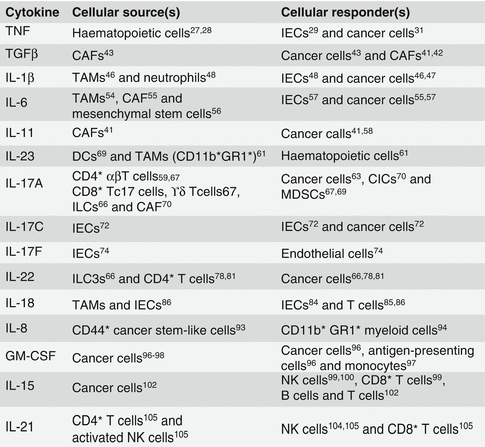
Table 7.1
JAK-STAT–dependent cytokine receptor systems implicated in solid malignancies
Cytokine | Cytokine receptors | JAK kinase | STAT |
|---|---|---|---|
IL-6 | IL-6R/sIL-6R/gp130 | JAK1, JAK2, TYK2 | STAT1, STAT2, STAT3, STAT4, STAT5 |
IL-4 | IL-4/13R | JAK1, JAK2, JAK3, TYK2 | STAT6 |
IL-10 | IL-10/20R | JAK1, JAK2, TYK2 | STAT1, STAT2, STAT3, STAT4, STAT5 |
IL-12 | IL-12Rβ1/β2 | JAK2, TYK2 | STAT3, STAT4 |
IL-23 | IL-23R/12Rβ1 | JAK2, TYK2 | STAT3, STAT4 |
GM-CSF | CD131 (βCR) | JAK2 | STAT3, STAT5, STAT6 |
Fig. 7.2
The role of cytokines in the development of colorectal cancer [184]. Cytokines produced by a variety of cell types, including innate and adaptive immune cells and tumour cells, play an important role in the development of colorectal cancer. Some cytokines can impair tumour growth via inhibition of angiogenesis and cancer dissemination. However, in well-established tumours, many cytokines can promote tumour proliferation, invasion, and metastasis. This schematic depicts where and how cytokines may influence the pathogenesis of colorectal cancer
Regulation of the JAK-STAT Pathway
Several intracellular components can regulate the activation of specific JAKs and STATs to modulate the central JAK-STAT signalling pathway. Certain signal-transducing adapter molecules (STAMs), including STAM1 and STAM2A, contain an inducible tyrosine-based activation motif that allows phosphorylation by JAK1 and JAK3 [147]. The STAMs facilitate transcriptional activation of specific target genes, although the mechanism by which this occurs is unclear.
Signalling adapter proteins, including LNK, SH2B, and APS, possess SH2 and Pleckstrin homology domains that modulate JAK2 activation [13]. LNK binds phosphorylated JAK2 and the product of a somatic mutation, JAK2V617F, to inhibit downstream signalling [22, 54], whereas SH2B and APS have been associated with promotion of JAK2 signalling.
The suppressor of cytokine signalling (SOCS) family functions in a negative feedback loop in which SOCS proteins are expressed downstream of JAK/STAT signalling and act as negative regulators of cytokine signalling [13]. Most SOCS members function as E3 ubiquitin ligases, promoting ubiquitination and degradation of JAK-associated cytokine receptors and possibly JAKs as well. SOCS-1 and SOCS-3 prevent downstream signalling via direct binding to JAK1, JAK2, and TYK2.
Protein tyrosine phosphatases (PTPs), such as shatterproof-1 (SHP-1), SHP-2, PTP1B, T-cell PTP, and CD45, bind to and dephosphorylate JAKs or cytokine receptors, thereby reversing JAK activity [13]. Protein inhibitors of activated STATs (PIAS) inhibit STAT signalling by blocking STAT DNA binding or by recruiting histone deacetylases or other corepressors [48]. Conversely, the methyltransferase enhancer of zeste homolog 2 has been reported to methylate STAT3, which confers enhanced STAT3 activity [90].
Lessons from JAK Knockout Mice: Role of JAK-STAT in Immune and Bone Marrow Function
JAK-STAT signalling is a key regulator of immune cell growth, survival, and differentiation in innate and adaptive immunity, as evidenced by data from murine models and clinical observations [55]. JAK1 and JAK2 deletions are lethal in mice as a result of nursing-related complications [126, 136, 149]. JAK3 and TYK2 deletions are not lethal, but significantly influence lymphocyte biology and cytokine signalling that is essential to the differentiation and function of T cells and other immune cells. In JAK1 −/− mice, a deficit in thymocyte production and maturation of B cells was observed, along with a lack of response of cells to interferon (IFN)-α and IFN-γ induction [149]. JAK3 knockout mice also have thymocyte deficiencies and profound reductions in mature B and T cells [129, 138, 169], although no apparent nonimmunologic deficits have been observed [169].
Interestingly, the immunodeficient phenotype of JAK3 knockout mice is similar to that of severe combined immunodeficiency syndrome in humans [129, 138, 169]. The effects of JAK3 deficiency on T-cell subsets in mice include an increase in the susceptibility of nature killer (NK) cells and intestinal γδ T cells to apoptosis, induction of peripheral CD4+ T-helper (Th) cell anergy, and inhibition of Th1/Th2 cell differentiation [55]. In addition, JAK3 knockout mice develop myeloid cell expansion and enhanced dendritic cell (DC) differentiation and cytokine production (e.g., interleukin [IL]-10, IL-12). TYK2 −/− mice differ from JAK3 −/− mice in that they show no obvious deficits in the numbers of lymphoid, myeloid, or monocyte cells [86, 157]. However, similar to JAK3 −/− mice, TYK2−/− mice also have impaired Th1 differentiation [133], which can be attributable to its essential role in IL-12 signalling [86, 157]. In addition, these mice are particularly susceptible to infection, suggesting that TYK2 plays an essential role in host defence [86]. For example, IL-23–induced IL-17 production by peritoneal γδ T cells was reduced in TYK2 −/− mice compared with wild-type mice, leading to impaired neutrophil infiltration and immune response against Escherichia coli infection [123]. Collectively, the data from JAK-STAT knockout studies confirm the importance of cytokines in regulating how the JAK-STAT pathway affects the immune system.
JAK-STAT Dysregulation in GI Cancers
JAK-STAT pathway dysregulation is an important mechanistic underpinning to the pathophysiology of many disorders (Fig. 7.3). Dysregulation of the JAK-STAT pathway is widely documented in GI cancers and may arise from cytokine-dependent or -independent mechanisms. Overexpression of cytokines resulting from uncontrolled immune and inflammatory stimuli in the tumour microenvironment can lead to overactivation of receptor-mediated JAK family members. Cytokine-independent mechanisms could include inactivation of negative regulators (e.g., SHPs, PIAS) or activating mutations in JAK family members. In particular, persistent STAT3 activation promotes multiple aspects of GI tumour development [25, 33, 127]. A recent meta-analysis of 22 studies (N = 3585 total patients) that evaluated the prognostic significance of phosphorylated STAT3 (pSTAT3) in GI cancers reported that pSTAT3 expression was significantly associated with shorter overall survival (OS; hazard ratio [HR], 1.809; P < 0.001), disease-free survival (DFS; HR, 1.481; P = 0.035), and other clinicopathological features, including tumour cell differentiation (odds ratio [OR], 1.895; P < 0.001) and lymph node metastases (OR, 2.108; P = 0.024) [109]. The precise aetiology and role of abnormal JAK-STAT signalling in individual cancers remains unclear [168]. However, a variety of data are available that implicate key JAK-STAT pathway components in multiple tumour-specific settings.
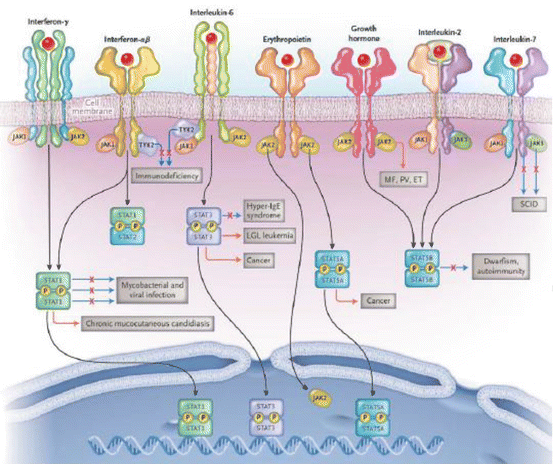
Fig. 7.3
Mutations of JAKs and STATs are associated with a variety of disorders [131]. Several JAK and STAT mutations result in dysregulation of the JAK-STAT signalling pathway and have been implicated in the pathogenesis of a variety of disorders. This schematic depicts where mutations in the JAK/STAT pathway can result in corresponding disorders
Colorectal Cancer
In colitis-associated cancer (CAC), various members of the JAK-STAT pathway have been associated with tumour promotion, progression, and metastasis, as well as antitumour activity [205]. In addition, overexpression of PIAS3 was observed in colorectal cancer (CRC) tissue compared with normal tissue [108]. An analysis of population-based case–control studies (N = 5224) showed that mutations in various JAKs and STATs were associated with increased mortality risks in patients with colon and rectal cancers [158]. However, other studies have reported contradicting prognostic evidence concerning pSTAT3 in CRC. In 104 French patients with advanced rectal cancer, pSTAT3 was an independent prognostic factor for improved OS (HR, 0.3; 95 % CI, 0.1–0.8; P = 0.01) [121]. In contrast, a study in 108 patients with colorectal adenocarcinoma reported that pSTAT3 expression was significantly correlated with shortened OS (P < 0.001) as well as with tumour invasion characteristics, including the depth grading of invasion, lymphatic invasion, and stages of Dukes’ classification [104]. These contrasting results may have arisen from differential STAT3 expression in specific cell types. For example, an immunogenic tumour containing STAT3-positive lymphocytes may be reflective of an appropriate antitumour immune response, resulting in improved patient survival. However, a tumour containing STAT3-positive nonimmune cells originating from either host or cancer cells may reflect a highly metastatic and invasive stage of tumour development. Given the broad tumour heterogeneity among patients with CRC, further studies are warranted to better characterise the cellular origin of JAK-STAT activation to understand of the role and clinical significance of JAKs and STATs in CRC.
Pancreatic Cancer
A variety of data suggest that STAT3 and IL-6 have important roles in the pathogenesis of pancreatic ductal adenocarcinoma (PDAC) [64]. Whereas inactive STAT3 is typical of nonmalignant pancreas tissue samples, activation of STAT3 via phosphorylation of tyrosine residue 705 has been observed in murine and human models of PDAC [64, 107]. IL-6, produced primarily by tumour-infiltrating macrophages, was demonstrated as the major driver of STAT3, the activity of which is mainly regulated by SOCS-3 in PDAC. In a study of transgenic mice, STAT3 was shown to induce the development of the acinar-to-ductal metaplasia via persistent expression of the transcription factor pancreatic and duodenal homeobox factor 1 (PDX-1) [120]. STAT3 is shown to be activated and overexpressed in ductal carcinoma cells compared with the ducts from chronic pancreatitis [153]. Functional inactivation of STAT3 in a subset of pancreatic cancer cell lines significantly inhibited cell proliferation in vitro and reduced tumour growth in vivo. In human pancreatic cancer, STAT3 has been shown to regulate vascular endothelial growth factor (VEGF) expression, angiogenesis, and metastasis [183], supporting a malignant phenotype of human pancreatic cancer.
Hepatocellular Carcinoma
Several JAK1 gene mutations, including S729C, N451S, E483D, and S703I, have been identified in tissues from patients with hepatocellular carcinoma (HCC) [84, 194]. However, only S729C and S703I are believed to confer a gain-of-function role in the JAK-STAT pathway. Cells with the S703I mutation were capable of continual proliferation and were sensitive to the JAK1/JAK2 inhibitor ruxolitinib [194]. In a study that examined the molecular pathogenesis of HCC, enhanced activation of JAK-STAT components, including JAK1, JAK2, TYK2, STAT1, STAT3, and STAT5, were observed in HCC tumour tissues (n = 80) compared with normal liver tissue (n = 10) [27, 156, 188]. Furthermore, hypermethylation of SOCS-3 promoters (i.e., SOCS-3 inactivation) was observed in a subgroup of patients with survival of <3 years. This is consistent with an analysis that found significant correlations between abnormal pSTAT and SOCS-3 expression patterns and survival [188]. Expression of the JAK-STAT negative modulator SOCS-1 was also found to be substantially reduced in samples from 4 of 8 HCC tumours tested in one analysis (Nagai et al. 2001), providing an additional mechanism for the overactivation of JAK-STAT signalling. As in pancreatic cancer settings, HCC tumourigenesis and proliferation has been shown to be closely associated with IL-6-STAT3 signalling [127].
Gastric Cancer
Activated STAT3 has been observed in GI stromal tumours with activating mutations in c-kit, along with reduced tumour cell growth with pharmacologic blockade of JAK2 [134]. Consistent with this finding, activated STAT3 was increased in excised gastric cancers compared with healthy adjacent tissue, and was associated with disease stage [52]. In a meta-analysis of studies that evaluated the prognostic significance of pSTAT3 in GI cancers, elevated pSTAT3 had the most notable association with worse OS among patients with gastric cancer (HR, 2.264; P < 0.001) compared with other malignancies of the digestive tracts, including oesophageal cancer (HR, 1.825; P = 0.092), pancreatic cancer (HR, 1.716; P = 0.196), HCC (HR, 1.654, P = 0.041), and CRC (HR, 1.149; P = 0.729) [109].
Involvement of Cytokines and Growth Factors That Signal Through the JAK-STAT Pathway in GI Cancers
The tumour microenvironment is regulated by cytokines, chemokines, growth factors, enzymes, and angiogenic mediators from myriad cell types, including immune cells, endothelial cells, mesenchymal cells, and tumour cells [144]. A variety of these mediators are involved in the complex tumour microenvironment. The serum and tumour expression patterns of key cytokines and granulocyte/macrophage colony-stimulating factor (GM-CSF) are summarised in Table 7.2 . This section discusses the key cytokines, including IL-6, IL-4, IL-10, IL-12, IL-23, and growth factor GM-CSF, that signal via the JAK-STAT pathway and are thought to play prominent roles in the pathogenesis in GI malignancies.
Table 7.2
Expression and roles of major cytokines in gastrointestinal cancers
Cytokine | Immune function | Inflammation | GI cancer | Expression and role |
|---|---|---|---|---|
IL-6 | Immune suppressing | Proinflammatory | Hepatocellular | |
Colorectal | Elevated serum expression [113], associated with tumour stage, NLR [93], and reductions in OS and DFS [190] Elevated expression in tumour tissue [34] | |||
Gastric | Elevated serum expression, associated with poor prognosis [113] | |||
Pancreatic | Promotes cancer development and progression [70] | |||
IL-4 | Immune suppressing | Proinflammatory | Hepatocellular | Elevated serum expression [180] |
Colorectal | Elevated expression in polyp versus normal epithelial tissue [164] Patients with elevated serum levels of sIL-4R were less likely to have ≥3 polyps [31] | |||
IL-10 | Immune suppressing | Anti-inflammatory | Hepatocellular | |
Colorectal | Elevated serum expression, associated with poor prognosis [113] | |||
Gastric | Elevated serum expression, associated with poor prognosis [113] | |||
Pancreatic | Elevated serum expression, associated with poor prognosis [113] | |||
IL-12 | Immune activating | Proinflammatory | Colorectal | Reduced serum expression [113] |
Gastric | Reduced serum expression [113] | |||
Hepatocellular | Reduced serum expression [113] | |||
IL-23 | Immune suppressing | Proinflammatory | Colorectal | Serum levels higher in early versus advance disease [177] |
Gastric | Elevated expression in tumour tissue [114] | |||
Pancreatic | ||||
GM-CSF | Immune activating Immune suppressing | Proinflammatory | Colon | |
Pancreatic | Elevated expression in tumour tissue [14] |
Interleukin-6
Interleukin-6 is a prototypic inflammatory cytokine that is produced by a variety of cell types, including hematopoietic cells, stromal cells, epithelial cells, and muscle cells. IL-6 can bind to corresponding membrane-bound receptors (IL-6R or CD126) or soluble receptors (sIL-6R). These complexes then bind to the gp130 IL-6 transducer (CD130), resulting in gp130 dimerisation, and phosphorylation and activation JAK1, JAK2, and Tyk2 [151]. Several studies have highlighted the effect of the IL-6/JAK/STAT signalling pathway on cancer initiation and progression, particularly in patients with CRC.
Increased expression of IL-6 in patients with CRC in serum and tumour tissue has been documented in a number of studies [51, 100]. In addition, IL-6 expression has been associated with increased tumour stage, size, and metastasis, and reduced survival in patients with CRC [96]. An association between serum levels of IL-6 and carcinoembryonic antigen has also been reported in patients with CRC [19]. Although data on IL-6 expression patterns in sporadic CRC are well established, the source of IL-6 expression in noninflammation–associated cancer is less clear. One explanation could be related to the infiltration of tumours with IL-6–secreting inflammatory cells as seen in patients with CAC.
Interleukin-6, secreted by lamina propria T cells and macrophages, was demonstrated to be important for the development in vivo in a murine model of CAC [16]. In the widely used mouse model of CAC using the mutagenic agents azoxymethane and dextran sulphate sodium, the authors observed intestinal tumour growth that was dependent on IL-6 trans-signalling in intestinal epithelial cells, possibly with downstream activation of STAT3. The tumour-promoting effect of IL-6 could be inhibited through treatment with anti–IL-6R antibodies or sgp130Fc, a designer variant of soluble gp130 that specifically blocks trans-signalling [16]. In addition, IL-6 was shown to mediate proliferative and antiapoptotic effects on malignant cells by activating STAT3 [58]. This is consistent with the finding that the majority of IL-6 target genes regulate cell cycle progression and the suppression of apoptosis [112]. During the acute inflammation phase, IL-6 was associated with immune response activation via recruitment of neutrophils and monocytes and subsequent transition to adaptive immunity via T-cell recruitment and differentiation towards Th2 and Th17 phenotypes [152].
Interleukin-6 has also been implicated in other inflammation-associated GI malignancies, including pancreatic cancer [201], HCC [81], and gastric cancer [181]. In patients with HCC, higher intratumoural Th17 cell density was associated with poor OS and DFS, suggesting that IL-6 may have immunosuppressive effects in GI malignancies [28, 198].
Interleukin-4
Interleukin-4 is a Th2 cell cytokine that regulates the immune response and microenvironment under normal physiological conditions and in cancer settings [162]. IL-4 binds to its high-affinity receptors on solid tumours, consisting of IL-4Rα and IL-13Rα1 chains, forming functional receptors in cancer cells that activate JAKs following ligand binding and heterodimerisation. Upon phosphorylation, 5 tyrosine residues (Y1–5) in the cytoplasmic tail of the IL-4Rα interact with signalling molecules to regulate the immune response through the JAK/STAT pathway [115]. Y1 is in the I4R sequence motif and interacts with the phosphotyrosine-binding domains of insulin receptor substrate 1 and 2. Y2–4 bind with the SH2 domain of STAT6, and Y5 is found in an ITIM consensus motif that brings SHP-1 to the activated receptor complex.
The biological effects of IL-4 in cancer cells include tumour proliferation, cell survival, cell adhesion, and metastasis. IL-4 is produced by several cell types, including mast cells, basophils, and activated T lymphocytes [110]. IL-4 is primarily known for regulating B and T lymphocytes [110]. Specifically, IL-4 can stimulate the proliferation and differentiation of these cell types and the expression of a variety of gene targets [110]. IL-4 has been shown to induce transient inflammatory responses in the colon tissue of mice via a STAT6-dependent mechanism [173]. Furthermore, IL-4 exerts carcinogenic effects that may be essential to the survival of colon cancer stem cells and mature cancer cells via STAT6-dependent regulation of antiapoptotic genes and related proteins (e.g., survivin) [37, 43]. This feature of IL-4 may also play a role in the chemoresistance mechanism observed in some patients with colon cancer via upregulation of proteins associated with multidrug resistance (e.g., P-glycoprotein) [43].
Endogenous IL-4 induces the immunosuppressive M2 phenotype of tumour-associated macrophages (TAMs) and Th2 differentiation [23, 203], which is often associated with poor prognosis in patients with GI malignancies [45]. Furthermore, IL-4 was shown to suppress the immune surveillance and support tumour growth and metastasis in patients with pancreatic cancer [142]. However, evidence suggests that exogenous IL-4 (i.e., not produced by immune cells) has an antitumour effect conferred in a biphasic fashion that involves innate immune cell accumulation and maturation, followed by CD8+ T-cell activation [110].
Interleukin-10
Interleukin-10 is a potent anti-inflammatory cytokine that signals through a variety of JAK-STAT signalling components, including JAK1, TYK2, and STAT3 [105, 112]. In patients with CRC, IL-10 serum levels increased over time during tumour progression [130], and high preoperative serum levels of IL-10 have correlated with poor survival (HR, 2.561; 95 % CI, 1.11–5.91; P = 0.023) [119]. These data suggest a tumour-promoting role for IL-10 in patients with CRC. However, IL-10 knockout mice were shown to be more susceptible to developing CAC than wild-type mice, whereas newborn IL-10 knockout mice treated with exogenous IL-10 showed no sign of intestinal inflammation or CAC [21]. Examples of the anti-inflammatory effects of IL-10 include downregulation of proinflammatory cytokine production by immune cells [11] and counteraction of IL-12–driven inflammation [69, 112]. In addition, IL-10 is an immunosuppressive cytokine that influences both the innate and adaptive immune responses [105]. In patients with HCC, IL-10 was shown to suppress antigen presentation, differentiation, and maturation of DCs, allowing tumour cells to evade immune surveillance mechanisms [15]. IL-10 has also been shown to help regulate the activities of regulatory T cells (Tregs) and the subsequent suppression of T-cell–mediated colitis and intestinal bowel disease, and prevent colon cancer [39, 116].
Interleukin-12 and Interleukin-23
Interleukin-12 and IL-23 are proinflammatory cytokines that have similar molecular characteristics, share a common subunit (IL-12p40), and signal via JAK2, TYK2, and a variety of STATs [112, 191]. IL-12 comprises the IL-12p40 subunit linked to the IL-12p35 subunit, which signals through the IL-12 receptor (IL-12R; includes β1 and β2 subunits) as a heterodimer. Upon binding to its receptor, IL-12 stimulates JAK2 and TYK2 activity, leading to phosphorylation of STAT1, STAT3, STAT5, and, in particular, STAT4 homodimers [182]. IL-23 comprises the IL-23p19 and the IL-12p40 (i.e., IL-12/23p40) subunits, which signal through IL-23R and IL-12Rβ1. Like IL-12, IL-23 activates JAK and STAT signalling molecules, predominantly STAT3. Despite their common features, IL-12 and IL-23 exhibit distinct effects on the immune response associated with cancer [191].
Endogenous IL-23 was suggested to support tumour growth, the expansion of Th17 cells, enhanced production of Th17 cell-related cytokines (e.g., IL-17), and reduced CD8+ T-cell infiltration [78, 106]. IL-23–deficient mice have demonstrated enhanced cytotoxic T-cell activity, which reduced tumour incidence and growth [106]. In mouse models of colon cancer, STAT3 activation in Tregs was shown to upregulate IL-23 and downregulate IL-12 expression [101]. Evidence suggests that dendritic cells, macrophages, and neutrophils produce IL-23 during intestinal inflammation [10, 94]. A variety of hematopoietic cells in the intestine, including innate lymphoid, Treg, and Th17 cells, can react to IL-23 [3]. The net biological effect of IL-23 signalling may indirectly promote tumour cell survival. IL-23 has been reported to drive intestinal inflammation by inducing other proinflammatory cytokines, such as IL-6, IL-17, and IL-22. These cytokines may in turn activate tumour cell proliferation through STAT3 and nuclear factor kappa B (NF-kB). IL-23 has been associated with traditional hallmarks of chronic inflammation in GI cancers, including enhanced activities of MMP9, angiogenesis, and macrophage infiltration [106].
The effects of IL-12 in the context of cancer are thought to generally be protective. Some data suggest that IL-12 promotes antitumour immunity via activation and proliferation of NK cells, Th1 cells, and cytotoxic T cells [112, 170]. In mouse models of colon carcinoma, IL-12 induced tumour suppression via an IFN-γ–independent fashion [170]. Interestingly, unlike IL-23, IL-12 expression was not elevated in human colon adenocarcinoma [106]. Clinical studies have attempted to evaluate the effects of recombinant IL-12 in patients with different cancer settings, including colon cancer, but meaningful results have not been observed [170].
Granulocyte/Macrophage Colony-Stimulating Factor
Granulocyte/macrophage colony-stimulating factor belongs to the hematopoietic growth factor family of cytokines and has significant functional homology with IL-3 and IL-5 related to a common β-receptor subunit [185]. The ligand specificity of GM-CSF is preserved by distinct α-receptor subunits. The α-subunits are specific for each cytokine and bind their specific ligand with low affinity. The βc-subunit forms a high-affinity receptor with all 3 α-subunits, despite its lack of capacity to bind the cytokines by itself. The βc-subunit is not only required for the formation of the high-affinity receptor complex but also crucial for signal transduction.
GM-CSF is elevated in the majority of PDAC patient samples. In a study evaluating the tumour tissue expression, 14 out of 16 patient samples were positive for GM-CSF (i.e., ≥75 % of pancreatic intraepithelial neoplasias within a section exhibited ≥50 % stained cells per lesion) [143]. Upregulated GM-CSF expression was also observed in invasive PDAC lesions, suggesting that overexpression of GM-CSF persists throughout disease progression. In addition, GM-CSF expression was not detected in lesions from non-PDAC cases, including chronic pancreatitis, pancreatic dermoid cyst, pancreatic endocrine neoplasm, and serous cystadenoma.
The effects of GM-CSF on the JAK-STAT signalling pathway are central to macrophage differentiation, tumour transformation via epithelial to mesenchymal transition, and tumour angiogenesis [4]. GM-CSF–activated STAT3 is involved in epithelial-mesenchymal transition regulation, and JAK2 and STAT3 are believed to play a role in blood vessel development [4]. In an elegant series of experiments using a genetically engineered mouse model of PDAC, two independent groups led by Drs. Pylaveya Gupta and Bayne demonstrated that Kras-induced GM-CSF production promotes the development of pancreatic neoplasia, promotes suppressive myeloid cells, and negatively modulates T-cell tumour immunity [14, 143]. In pancreatic ductal cells harbouring oncogenic Kras, GM-CSF production is associated with the expansion of a heterogenous Gr1+CD11b+ myeloid cell population, including myeloid-derived suppressor cells, monocytes, and immature myeloid cells [143]. Several lines of evidence suggest that Gr1+CD11b+ cells are immunosuppressive. Of particular relevance is that tumour cell-derived GM-CSF can orchestrate an immunosuppressive crosstalk between Gr1+ CD11b+ immature myeloid cells and CD8+ T cells [14]. Abrogation of tumour-derived GM-CSF led to reduced Gr-1+ CD11b+ cell infiltration and failure of implanted tumours to grow; however, CD8+ T-cell depletion was able to restore tumour growth. The potential relevance of this immune modulatory mechanism to advanced stages of pancreatic cancer is further supported by the reciprocal relationship between CD8+ T cells and Gr1+CD11b+ cell infiltrates observed in pancreatic tumours from the murine PDAC model.
The role of GM-CSF in other GI malignancies is not well characterised and may be multifactorial. In CRC, GM-CSF has been proposed to induce tumour suppression via separate immune-mediated and immune-independent pathways [172]; abolishing GM-CSF resulted in marked increases in tumour volume and weight in vivo. In addition, T-lymphocyte infiltration and T-cell–mediated cytolytic effects were higher in GM-CSF–expressing tumours compared with controls [172], suggesting that findings in PDAC may not be applicable to other GI cancer settings. Further studies are needed in carefully controlled experiments utilising patient-derived tissues to determine the clinical settings in which GM-CSF blockade or addition could be beneficial.
Regulation of the Tumour Microenvironment by JAK-Signalling Cytokines
Bone marrow-derived cells of the innate and adaptive immune systems are recruited to the tumour microenvironment during the inflammatory response and may play different roles in promoting or suppressing GI carcinogenesis [41, 60, 144]. JAK-signalling cytokines represent a key mechanism by which immune and inflammatory cells interact with the tumour and tumour stroma to influence tumour initiation, promotion, and metastasis (Fig. 7.4). Individual cellular components are explored in this section to further understand the potential impact of JAK-STAT modulation on the tumour microenvironment.
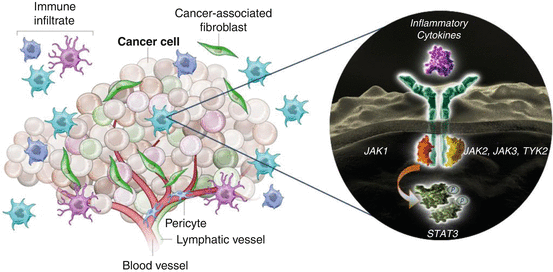
Fig. 7.4
Tumour-associated inflammation orchestrated by cytokines and JAK-STAT signalling (Juntila 2013). The JAK-STAT signalling pathway is central to signal transduction of inflammatory cytokines in the tumour microenvironment and the regulation of tumour-associated inflammation, including immune cell infiltration and new blood vessel development
Inflammatory Component of GI Tumours
Tumour-associated inflammation is an important component of oncogenesis in a variety of cancers [62]. Several types of inflammatory cells infiltrate preceding or following tumour formation and may be induced by cancer treatments, including chemotherapy and radiation [60]. Many types of GI malignancies, including CRC, pancreatic cancer, HCC, and gastric cancer, may arise from chronic inflammatory disorders [145]. In CRC, inflammatory signals (e.g., via IL-6), are essential to the transformation of adenomas to carcinomas via activation of antiapoptotic and proliferative factors, including STAT3 [59]. The activation of several oncogenes, including KRAS, p53, APC, and β-catenin, were suggested to promote inflammation in GI malignancies, primarily through dysregulated NF-kB and STAT3 signalling [38, 40, 167].
In the microenvironment of solid tumours, including GI malignancies, inflammation is involved in complex mechanisms that directly and indirectly affect multiple aspects of tumour biology, including tumour initiation, tumour development, and angiogenesis [60]. A variety of evidence suggests that tumourigenesis and inflammation operate in tandem. Reactive nitrogen and oxygen species and tumour-associated cytokines produced from activated inflammatory cells may induce DNA damage and epigenetic changes, including stem cell-like phenotypes in tumour progenitor cells, which contribute to the genetic instability that promotes tumour initiation [60, 89, 146]. In addition, chronic inflammation can perpetuate this instability via diminished DNA mismatch repair mechanisms, DNA damage-induced apoptosis, and cell-cycle control [89, 146]. These changes can induce proinflammatory cytokine production and further inflammatory responses [60].
Cytokines and enzymes produced by inflammatory cells in the GI tumour microenvironment also support tumour survival, proliferation, invasion, and metastasis, which are primarily regulated via NF-kB and STAT3 signalling pathways [61]. For example, the proinflammatory cytokine IL-6 was demonstrated to regulate the survival of malignant pancreatic cells via upregulation of antiapoptotic genes, including BcL-2 and BcL-xL, and the proliferation of pancreatic cancer cells was shown to be regulated via IL-4, IL-6, and IL-8. Studies have also demonstrated that other proinflammatory cytokines (e.g., IL-1α, IL-1β, transforming growth factor beta [TGF-β]) play important roles in the invasive and metastatic mechanisms of pancreatic cancer cells. In a model of colon cancer, matrix metalloproteinases 2 and 9 (MMP2, MMP9) produced by immature myeloid cells (iMCs) were also associated with tumour invasiveness [95].
The “angiogenic switch,” or promotion of intratumoural blood supply in solid tumours, including GI malignancies, is essential for tumourigenesis and can be turned on via tumour-associated inflammatory responses [102]. Triggered by a hypoxic tumour microenvironment, hypoxia-inducible factor-1α upregulates the expression of different proangiogenic factors, including VEGF, angiopoietin, and other proinflammatory cytokines/chemokines [102]. In pancreatic cancer, the angiogenic effect is primarily mediated via proinflammatory IL-6 and IL-8 signalling [64].
Triggers of inflammation in GI cancers can also be indirect, stemming from environmental or infectious origins. For example, associations between infection and risk of cancers have been observed between hepatitis B and C viruses and HCC and between Schistosoma or Bacteroides species and colon cancer [60]. In addition, obesity has been proposed to induce HCC and pancreatic cancer in some patients because of obesity-related chronic inflammation [88, 137]. Finally, inflammatory cytokines and growth factors, including cyclooxygenase-2 and VEGF, have been described as components of the mechanisms through which nicotine promotes GI carcinogenesis [80].
Innate Immune Components of GI Cancers: Myeloid Cells
Myeloid-derived suppressor cells (MDSCs), TAMs, and tumour-associated neutrophils (TANs) are among the most abundant cells of the innate immune system in the tumour microenvironment [144, 175]. MDSCs consist of a heterogenous population of activated iMCs that are produced excessively in tumour-bearing mice and human cancer settings [144]. Significant tumour infiltration of iMCs in mouse models of colon cancer and oesophageal squamous cancer suggest that MDSCs are implicated in the pathogenesis of GI malignancies [160, 165]. Under pathological conditions, components of the tumour microenvironment alter the normal differentiation process by expanding and activating iMCs to MDSCs [49]. A variety of cytokines and growth factors can utilise the JAK-STAT signalling pathway to induce MDSC expansion and activation in GI malignancies, including VEGF, GM-CSF, IFN-γ, IL-4, IL-10, IL-12, and IL-13. JAK2/TYK2-STAT3, JAK1/JAK3-STAT6, and JAK1-STAT1 are major components of signalling pathways involved in the expansion and activation of MDSC populations. Upon activation, MDSCs produce immunosuppressive factors, including TGF-β, IL-10, IL-12, arginase, nitric oxide synthase (iNOS or NOS2), nitric oxide, and reactive oxygen species that inhibit cytotoxic T cells, NK cells, and DCs while promoting Th2 and Treg cells [49, 68, 144, 195].
Under specific chemokine/cytokine and growth factor signalling, peripheral blood monocytes are recruited and differentiated into macrophages with two principal phenotypes, the classically activated M1 and alternatively activated M2 [23, 91]. IL4/IL-13 and IL-10 activate STAT6 and STAT3, respectively, and induce M2 and M2-like phenotype, whereas IFN-γ and lipopolysaccharide activate STAT1 and other transcription factors that lead to M1 phenotype polarisation [23]. The different roles of M1 and M2 in carcinogenesis and inflammation are also dependent on the types of cytokines and chemokines they express [50]. M1 macrophages are typically characterised by a low expression of immunosuppressive IL-10, a high expression of IL-12 and Th1-attractant chemokines (e.g., CXCL9, CXCL10), and the production of proinflammatory cytokines (e.g., IL-12, IL-23). Therefore, the M1 phenotype has been proposed to play a crucial role in enhancing Th1 responses and mediating cytotoxic activities against tumours [50]. In contrast, M2 macrophages are characterised by elevated IL-10 levels and other molecules involved in the recruitment of Th2, Treg, and other immune cells that promote tumour growth, invasion, and metastasis, as well as angiogenesis, while inhibiting T-cell–mediated antitumour activities [50]. In addition, the M2 phenotype has been associated with inflammation resolution [23].
Tumour-associated neutrophils are mature neutrophils in the tumour microenvironment that are different from their granulocyte precursors, as evidenced by findings from transcriptomic analyses [44, 91]. Emerging evidence has suggested that neutrophils participate in both procarcinogenic and anticarcinogenic processes via specific cytokines and other mediating molecules in different types of cancers, including GI malignancies [50]. The combination of IL-6 and G-CSF was shown to augment STAT3 signalling in neutrophils in vivo and may promote tumour growth and angiogenesis [193]. Similar to the phenotype polarisation observed with macrophages, TANs may adopt either an N1 or N2 phenotype, with a skewed differentiation towards the latter [44]. The proinflammatory N1 phenotype was proposed to amplify intratumoural CD8+ T-cell activities via production of T-cell–attracting cytokines (CXCL9, CXCL10, CCL3) and proinflammatory cytokines (IL-12, TNF-α, GM-CSF, and VEGF) [44]. In contrast, N2 neutrophils suppress T-cell–effector activities via production of high arginase levels and do not promote inflammation [44]. Findings from a mouse model of colon carcinoma indicated that CD8+ T-cell depletion led to decreased TANs in the tumour microenvironment, suggesting a possible mechanism by which T cells may attract and activate TANs [44].
Both the antitumour immunity and tumour-promoting effects of DCs have been implicated in the pathogenesis of solid tumours, including GI malignancies [30]. At the interface of innate and adaptive immune systems, increased tumour-infiltrating DCs was correlated with increased infiltration of CD8+ and CD4+ lymphocyte subsets, highlighting the essential role of DCs in antitumour activities via recruitment of cytotoxic T cells [35]. However, it was demonstrated that tumour-derived factors, including IL-4 and GM-CSF, activated the JAK2/STAT3 signalling pathway in iMCs and impaired the differentiation process of DCs, which could potentiate immune evasion [125]. In vitro and in vivo studies in human pancreatic carcinoma cells demonstrated that tumour-derived cytokines, including TGF-β, IL-10, and IL-6, reduced DC survival and proliferation and promoted differentiation toward the immunosuppressive plasmatoid DC phenotype versus the protective myeloid phenotype [17]. The immunosuppressive and procarcinogenic effects of tumour-associated DCs are primarily dependent on the production indoleamine 2,3-dioxygenase enzymes, which inhibit effector T cells; TGF-β, which expands Tregs; and angiogenic factors that promote new blood vessel development in the tumour microenvironment [20, 30].
Adaptive Immune Components of GI Cancers: Lymphoid Cells
Lymphoid-derived cells, including T-cell subsets, are members of the adaptive immune system and have essential roles in the tumour microenvironment [60]. Several T-cell subtypes are present in the tumour microenvironment and play an important role in carcinogenesis, including CD8+ cytotoxic T cells, CD4+ Th cells, Tregs, and NK cells [45, 60]. A T-cell–mediated adaptive immune response is a key driver of antitumour activities, primarily via cytotoxic and Th cells [5].
The differentiation of CD4+-naive T cells into Th1, Th2, Th17, or Treg cells depends on the cytokine environment and the corresponding affect on STAT proteins [203]. IL-12 and IFN-γ induce Th1 differentiation via STAT4 and STAT1/2 heterodimers, whereas IL-4 and IL-2 induce the Th2 phenotype via STAT5 and STAT6 [203]. Th cells can mediate an antitumour response through multiple mechanisms, including the regulation of adaptive and innate immune cells [97]. They can directly or indirectly enhance cytotoxic T-cell growth through IL-2 signalling, activate APCs, and reactivate memory T cells. Cytokines produced by Th cells can also recruit macrophages and eosinophils to the tumour site, which produce cytotoxic factors, including superoxide and nitric oxide [73]. In GI malignancies, Th2 cells can induce inflammation and support tumour growth [144]. In murine models of colitis-associated colon cancer, IL-4 produced by Th2 was found to induce tumour proliferation [99]. The signature cytokines of Th2, including IL-4 and IL-13, can also contribute to tumour formation and progression via M2 phenotype induction of TAMs [23].
Major JAK signalling cytokines involved in T-cell differentiation include IL-6, IL-21, and IL-23. All 3 of these cytokines have been shown to activate STAT3 and induce Th17 development, whereas Treg development is dependent on IL-2–driven STAT5 activation [203]. In addition to being induced locally, Tregs can also be recruited from peripheral organs to the tumour microenvironment [45, 204]. Studies have demonstrated elevated Th17 cell levels in the peripheral blood of patients with gastric and pancreatic cancer, suggesting a role in carcinogenesis [103, 197]. Th17 cells also produce a wide array of other cytokines, including TNF-α, IL-21, IL-22, GM-CSF, and IFN-γ, that can regulate the tumour microenvironment [6]. In patients with early-stage GI cancers, Th17 and Treg-cell densities were elevated in the tumour microenvironment compared with intraepithelial lymphocytes from healthy gastric mucosa [118]. Interestingly, as the tumours progressed, a decrease in Th17 cells and an increase in Treg cells was observed [118]. This observation is consistent with the hypothesis that Tregs suppress adaptive and innate antitumour immunity to enable tumour evasion [5, 60, 116]. Multiple suppressive mechanisms of Tregs have been described, including inhibition of effector T-cell proliferation and APC function in a juxtacrine or paracrine fashion [5, 204] and suppression of antitumour activities via the production of immunosuppressive cytokines [176].
NK cells are a subset of T cells that are known for their direct and indirect antitumour effects via activation of tumour cell apoptosis or induction of Th cells [175]. Nearly all aspects of NK cell biology are dependent on a core set of cytokines and associated receptors that signal via virtually all components of the JAK-STAT pathway, including IL-2, IL-12, IL-15, IL-18, and IL-21 [150]. Several clinical trials have evaluated these cytokines as therapeutic targets in the context of GI malignancies, including IL-12 in colon cancer and IL-21 in CRC. NK cells also produce a mixture of cytokines, including IFN-γ, that promote a Th1 response [57]. Other chemokines produced by NK cells can also promote the antigen-presenting function of other immune cells in the inflammatory tumour microenvironment [57]. However, these functions can be dysregulated by immunosuppressive cells, including MDSCs, TAMs, Tregs, and tumour-associated fibroblasts, and associated mediators that exist in the tumour microenvironment [175].
Therapeutic Approaches to Modulate JAK-STAT Pathway for the Treatment of GI Cancers
Overactive JAK-STAT signalling [156] resulting from tumour intrinsic or extrinsic mechanisms, gain of function mutations, or abnormal cytokine stimulation has important tumour-promoting effects, including tumour cell proliferation, invasiveness, metastatic potential, and antiapoptotic effects. Given the success of treatments targeting mutated kinases (e.g., Bcr-Abl, epidermal growth factor receptor [EGFR], B-raf, ALK, or those that are less critical for normal immune functions, such as VEGF receptor kinase, BTK, and PDGF), it is tempting to approach JAK inhibitors in a similar manner (i.e., maximal and around-the-clock inhibition). However, considering the critical roles that the JAK-STAT pathway plays in regulating normal physiological processes (e.g., immune and bone marrow functions), the goal of JAK-targeted therapies should be to achieve normalisation rather than complete inhibition. To achieve this goal, the use of highly selective inhibitors with well-defined dosing regimens is required to limit treatment-related side effects while maximising efficacy.
This section considers the published preclinical and clinical data regarding the activity of small molecule inhibitors of JAK-STAT signalling in GI cancers and also explores the potential to combine JAK-STAT–targeted therapies with immune checkpoint inhibitors for the development of optimised, comprehensive immunomodulatory treatment approaches for GI cancers.
Small-Molecule JAK or STAT Inhibitors
Several direct inhibitors of JAKs and STATs have been evaluated for a variety of disorders, but only a limited number of trials have included patients with GI cancers. However, existing preclinical evidence support antitumour activity for JAK inhibitors in models for CRC [8, 155, 178], pancreatic cancer [56, 72, 166], HCC [47, 187], and gastric cancer [83].
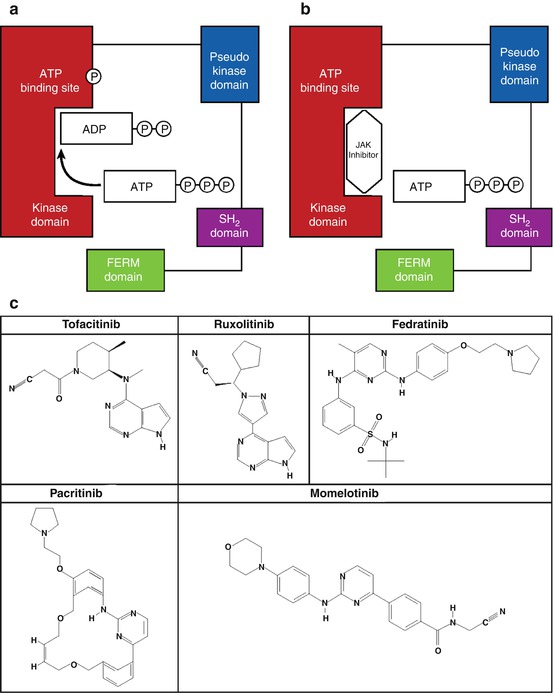
Typical JAK inhibitors are ATP mimetics that bind to the catalytic domain of the enzyme and competitively inhibit ATP from binding, thereby inhibiting the phosphotransfer reaction (Fig. 7.5). Tofacitinib (Xeljanz®, Pfizer Inc, NY, NY) [189] and ruxolitinib (Jakafi®, Incyte Corporation, Wilmington, DE) [79] are 2 JAK inhibitors that have been approved by the US Food and Drug Administration (FDA) for use in humans. In addition, several other JAK inhibitors are being evaluated in ongoing trials. The effectiveness of these inhibitors in patients with GI cancer remains unclear. Tofacitinib has inhibitory activity against JAK1, JAK2, and, JAK3 and is approved for patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate Tofacitinib is also being tested for other autoimmune conditions, such as alopecia areata, atopic dermatitis, psoriasis and psoriatic arthritis, ankylosing spondylitis, systemic lupus erythematosus, ulcerative colitis, and keratoconjunctivitis sicca; there is no evidence, however, of ongoing trials in patients with cancer.