The Mucosal Immune System
Yasmine Belkaid
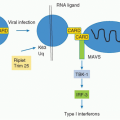
INTRODUCTION
Most antigens encountered by the immune system enter the body through the mucosal surfaces of the respiratory, gastrointestinal (GI), and urogenital tracts, and the vast majority of infectious microorganisms use the mucosae as portals of entry. These sites are, therefore, charged with the formidable task of protecting the host from environmental and pathogenic challenges while preserving vital physiologic tissue functions. The GI tract in particular represents the most reactive and complex immune environment of the host. Along with its constant exposure to food antigen and primary role in acquisition of metabolites, gut mucosal surfaces host complex microbial communities whose combined membership outnumbers host somatic cells. This enormous and highly variable antigenic load presents a significant challenge for the host immune system. When uncontrolled, reactivity against innocuous antigens such as those derived from food and intestinal flora poses a substantial risk that can lead to tissue damage and severe inflammatory disorders. Therefore, multiple highly specialized innate and adaptive immune cell types, as well as structural features, have been put in place to prevent overt reactivity and favor the induction of tolerogenic responses. However, tolerance is not the only fate of the immune response at mucosal sites as a certain degree of constitutive activation and inflammation is beneficial for the host, not only to reinforce the barrier, but also to allow for the development of protective responses when required. Indeed, the majority of infectious diseases worldwide either target or are acquired through mucosal surfaces of the GI, respiratory, and genital tracts. The complex and highly dynamic maintenance of immune tolerance to mucosal antigens concomitantly with the induction of protective responses to pathogens requires an arsenal of unique cells specifically conditioned by the mucosal environment. Some of the main actors in this control are unique antigen-presenting cells (APCs), various regulatory T (Treg) cell populations, intraepithelial lymphocytes (IELs), as well as recently identified innate lymphoid cells (ILCs). Further, the mucosal environment is under dominant control by the microbiota and dietary metabolites that direct the development and function of both innate and adaptive mucosal responses.
MUCOSAL COMPARTMENTALIZATION
The Gut-Associated Lymphoid Tissue
The mucosal immune system of the GI tract contains unique cell subsets that are structured in such a way as to favor the coexistence of the host with its flora, support food absorption, as well as induction of effector responses to pathogens.1 The primary barrier preventing transit of microorganisms and large molecules from the environment into the body is a simple epithelium made up of a single layer of cells overlaid with mucus and connected by junctional complexes. The gutassociated lymphoid tissue (GALT) can be divided into effector sites consisting of lymphocytes scattered throughout the epithelium and lamina propria (LP) of the mucosa and organized inductive sites. These inductive sites include the largest lymph node of the body, the mesenteric lymph node (MLN), Peyer patches, as well as cryptopatches and isolated lymphoid follicles (ILFs) that are distributed along the wall of the small and large intestine.1 ILFs are inducible structures recently described in both mice and humans that develop after birth in response to environmental signals, such as those derived form the microbiota.2,3,4 Peyer patches are macroscopic lymphoid aggregates consisting of large B-cell follicles with interspersed T-cell areas found in the submucosa along the length of the small intestine. In these structures, the lymphoid areas are separated from the intestinal lumen by a single layer of epithelial cells known as the follicle-associated epithelium and a more diffuse area below the epithelium known as the subepithelial dome. Notably, the follicle-associated epithelium contains unique cells referred to as microfold cells that have the capacity to transport organisms and particles from the gut lumen across the epithelial barrier.5 These cells also represent a major point of pathogen entry.5
Antimicrobial Peptides
A central strategy utilized by the mucosal immune system to maintain its homeostatic relationship with the microbiota and limit pathogen exposure is to minimize contact between luminal microorganisms and the epithelial cell surface. This is accomplished by promoting a physical barrier through the production of mucus, immunoglobulin (Ig)A, and antimicrobial proteins. All intestinal cell lineages, including enterocytes, goblet cells, and Paneth cells, can produce antimicrobial peptides. These molecules play a significant role in the control of pathogenic infections as well as in limiting exposure to the commensal microbiota. Epithelial cell-derived antimicrobial proteins are members of a diverse family including defensins, cathelicidins, and C-type lectins.6 Some of these molecules, such as α-defensins, are constitutively expressed. In other cases, engagement of pattern recognition receptors (PRRs) by commensally derived products induce the expression of a variety of antimicrobial peptides, which are critical to prevent translocation of gut bacteria across mucosal barriers.6 These proteins can exert antimicrobial functions by direct killing resulting from enzymatic attack of the bacterial cell wall or by
disrupting the bacterial inner membrane.6 Additionally, some antimicrobial proteins can function by depriving bacteria of essential heavy metals, such as iron.7 One of the best characterized mucosal antimicrobial peptides is RegIIIγ. This lectin is expressed soon after birth or following colonization of germfree mice.8 Production of RegIIIγ is tightly controlled by the flora in an MyD88-dependent manner and has a direct microbicidal effect on gram-positive bacteria.8,9,10 Similarly, the PRR nucleotide-binding oligomerization domain-containing protein (NOD) 2 controls expression of a subset of α-defensins and cryptdins by Paneth cells.11 Critically, antimicrobial proteins are retained in the mucus layer and are virtually absent from the luminal content.12 Such a feature allows antimicrobial activity to be concentrated within a region bordering the epithelial cell layer. Recent findings demonstrate that the accumulation of RegIIIγ in the mucus contributes to the maintenance of the segregation between the microbiota and the host intestine.13
disrupting the bacterial inner membrane.6 Additionally, some antimicrobial proteins can function by depriving bacteria of essential heavy metals, such as iron.7 One of the best characterized mucosal antimicrobial peptides is RegIIIγ. This lectin is expressed soon after birth or following colonization of germfree mice.8 Production of RegIIIγ is tightly controlled by the flora in an MyD88-dependent manner and has a direct microbicidal effect on gram-positive bacteria.8,9,10 Similarly, the PRR nucleotide-binding oligomerization domain-containing protein (NOD) 2 controls expression of a subset of α-defensins and cryptdins by Paneth cells.11 Critically, antimicrobial proteins are retained in the mucus layer and are virtually absent from the luminal content.12 Such a feature allows antimicrobial activity to be concentrated within a region bordering the epithelial cell layer. Recent findings demonstrate that the accumulation of RegIIIγ in the mucus contributes to the maintenance of the segregation between the microbiota and the host intestine.13
Mucus
The stratified mucus layer plays a crucial role in the maintenance of the physical segregation between the microbiota and host tissue, as well as preventing pathogen invasion and supporting their clearance. In particular, the mucus layer limits commensal and pathogenic bacterial contact with the epithelium preventing transport of bacteria to distal sites.14 To date,17 mucus (MUC) genes have been described,7 of them coding for secreted mucins (MUC2, MUC5A, MUC5B, MUC6, MUCC19, MUC7, and MUC8) that are differentially expressed in various mucosal compartments.14 The secreted, gel-forming, mucin glycoproteins that constitute the major macromolecular component of mucus are produced by goblet cells found throughout the GI tract and airways. In the airways, both mucus cells and serous cells produce defined mucins.15 Paneth cells represent another major secretory cell type within the GI tract.14 Mucus is found in the entire GI tract but varies in thickness ranging from 700 µm in the stomach and large intestine to 150 to 300 µm in the small intestine.16 The inner layer of the colonic mucus is attached to the epithelium and has a stratified appearance, ranging in thickness from 50 to 100 µm. Based on its structural capacity, this layer acts as a filter and is impermeable to commensal bacteria.17,18,19 The inner mucus layer is renewed by goblet cell secretion and at its luminal border is converted into an outer mucus layer. This outer layer expands in volume allowing commensal bacteria access to this zone.18 Contrasting the large intestine, the small intestine is only covered by a single layer of mucus that is not attached to the epithelium and more permeable to bacteria.16,20 Of note, commensal bacteria have a high proportion of their genome devoted to production of enzymes involved in glycan degradation and are able to utilize the released mucin monosaccharides as energy sources.21,22
MUCOSAL ANTIBODIES
About three-quarters of total antibody production is composed of IgA, in the order of 3 to 5 g produced per day in humans, with the vast majority secreted across barrier surfaces.23,24,25 In contrast, IgG is dominant in urine, bile, genital, and bronchoalveolar secretions; IgD is detected in nasal lacrimal as well as bronchoalveolar secretions,26,27,28; and IgE in nasal bronchoalveolar and intestinal secretions in the context of allergy.29 Human B cells produce two IgA subclasses, IgA1 and IgA2, whereas mice only produce one kind of IgA.30,31 IgA1 can be detected at systemic and mucosal sites, whereas IgA2 is essentially only present at mucosal sites.32 IgA can exist in a dimeric form called secretory IgA that is the main Ig found in mucus secretions, including tears, saliva, colostrum, and secretions from the genitourinary tract, GI tract, prostate, and respiratory epithelium.23,24,25 IgA is a major factor involved in the host-microbe dialogue at mucosal sites. It is critically important for shaping the microbiota, mediating pathogen clearance, neutralizing toxins and inflammatory microbial molecules such as lipopolysaccharides, as well as preventing adhesion of commensal bacteria to the epithelial surfaces by generating steric hindrance.30,31 Moreover, it can also induce bacterial agglutination and interact with the mucus layer through secretory component.33,34,35,36,37,38 One remarkable feature of IgA is its capacity to disseminate between mucosa-associated lymphoid structures. Such a feature allows specificities of IgA present in breast milk to be matched to the dominant microbes resident in the maternal microbiota.39,40 IgA induction is exquisitely sensitive to the presence of commensal bacteria in the intestine, and germ-free mice have very low levels of IgA that can be restored by commensal colonization.41,42,43,44
Immunoglobulin A Generation and Secretion
More than 40 years ago, Peyer patches were identified as the major source of IgA-producing B cells.16 IgA can also be generated in regional lymph nodes as well as in ILFs.45 The diffuse tissue of the LP can also support IgA production in the absence of follicular structures.46,47,48 In order to express IgA, B cells must undergo class switch recombination in which deoxyribonucleic acid (DNA) is spliced out of the heavy chain constant region loci to replace the µ/δ genes with the α gene segment downstream of the recombined VDJ segment. The GC-rich splice sites upstream of the gene segment for each Ig constant region are the targets for enzymes such as activation-induced (cytidine) deaminase (AID), which catalyze the reaction sequence. In humans, there are two Cα regions generating the two IgA isotypes, IgA1 and IgA2.30,31,32 Following induction in the intestinal lymphoid structures, B cells recirculate through the mesentery, lymphatic systems, and the bloodstream to home back to the intestinal mucosa where they develop into IgA-secreting plasma cells. Secretory IgA is produced as a result of cooperation between plasma cells in the LP that secrete dimeric IgA and epithelial cells that transport IgA into the lumen. This mechanism of transport involves covalent binding of dimeric IgA to the polymeric Ig receptor, which then carries it within vesicles through intestinal epithelial cells to the luminal surface. At this point, IgA is released by proteolytic cleavage of the polymeric Ig receptor leaving a fragment of the transport protein (termed secretory component) that remains associated with the joining chain of the IgA dimer forming a secretory IgA complex with noninflammatory protective functions.49,50,51 Indeed, secretory IgA can bind to bacteria without activating complement or stimulating the release of inflammatory mediators by innate cells.52,53
T-Dependent and -Independent Immunoglobulin A Generation
Mucosal IgA can recognize antigen with high- and lowaffinity binding modes.54 Generally, high-affinity IgA neutralizes microbial toxins and invasive pathogens whereas low-affinity IgA confines commensals to the lumen.32 Highaffinity IgA is believed to emerge from follicular B cells stimulated in a T cell-dependent manner, whereas low-affinity IgA can develop from extrafollicular B cells stimulated in a T cell-independent fashion.55 However, such distinction of origin and function is not absolute.56 Antigen at mucosal sites can stimulate IgA responses through T cell-dependent pathways that are initiated in mucosal lymphoid structures such as Peyer patches and MLN.54
Various mechanisms can promote the induction of IgA in a T cell-independent manner. In particular, transforming growth factor (TGF)-β that is highly enriched at mucosal surfaces controls B-cell responsiveness and promotes class switch recombination to IgA, in combination with interleukin (IL)-2 and IL-10 that act synergistically.57 Mechanistically, TGF-β1 binds to the TGF-β receptor complex that has a sterile transcript for the Iα locus upstream of the Sα recombination segment. Such a phenomenon is associated with signaling via mothers against decapentaplegic-2, -3, and -4 that bind to the Iα promoter in combination with the runt DNA binding factor RUNX3.58,59,60,61,62,63,64 A cytokine of the tumor necrosis factor (TNF) ligand family referred to as B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL) can also induce NF-κB activation and IgA induction.65,66 Further, as will be described in more detail later in this chapter, synthesis of the vitamin A metabolite retinoic acid (RA) by GALT dendritic cells (DCs) is crucial for the generation of mucosal IgA.67,68
Immunoglobulin A in Host Microbe Interaction
IgA plays a major homeostatic role by controlling host interactions with the microbiota as well as a protective role against pathogens.6,69 Although nonspecific IgA can limit epithelial invasion, pathogen-specific IgA plays an important role in the control of mucosal infections. For example, IgA production has been shown to control Shigella flexneri during infection by binding the bacteria at the mucosal layer,37 whereas IgA against the O-antigen component of lipopolysaccharides can protect against Salmonella infection.70 IgA can also shape the microbiota composition, as demonstrated in mice lacking IgA that have expansion of segmented filamentous bacteria (SFB) and other Clostridium-related species in the intestinal tract.71 In the context of IgA-microbiota dialogue, IgA responses lack typical memory characteristics and respond rapidly to changes in gut flora composition. This point was demonstrated using reversible colonization of mice with a strain of auxotrophic Escherichia coli that required nutrients unavailable from mammalian host metabolites.72 IgA responses persisted in the absence of bacteria; however, introduction of additional species of bacteria caused a rapid decline in IgA specific for the auxotrophic E. coli.72 As the microbiota composition can fluctuate in the context of infection or due to dietary changes, such a strategy would promote a dynamic IgA repertoire, allowing IgA to swiftly adapt to the commensals present at a given time.72
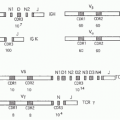
TABLE 34.1 Subsets of Intraepithelial Lymphocytes in Mice and Humans | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
INTRAEPITHELIAL LYMPHOCYTES
IELs reside within the epithelial layer and represent a highly heterogeneous group of cells distributed along the GI tract73 (Table 34.1). IELs represent one of the most abundant T-cell populations, and it has been estimated that 1 IEL is present for every 4 to 10 epithelial cells in the small intestine and 30 to 50 in the large intestine.74 These cells are important in the induction of both protective and regulatory responses at mucosal sites. They express features of adaptive and innate immune cells that allow them to survey tissue based not only on antigen recognition but also stress signals.73 IELs can be divided based on their expression of αβ and γδ T-cell receptors (TCRs) and further subdivided on the basis of
their cluster of differentiation (CD)8 expression with a large fraction expressing CD8αα (see Table 34.1).75
their cluster of differentiation (CD)8 expression with a large fraction expressing CD8αα (see Table 34.1).75
Homeostasis of Intraepithelial Lymphocytes
In the small intestine, IELs are highly abundant and are essentially composed of TCRαβ+ or TCRγδ+ CD8+ T cells. In contrast, in the large intestine the frequency of IELs is reduced and mostly represented by TCRαβ CD4+ T cells.74,76,77,78 Although the development and origin of the various IELs is the subject of long debate, all IEL subsets are progeny of bone marrow precursors that initially develop in the thymus.79 Some IEL precursors go through alternative self-antigen-based thymic maturation processes that lead to the functional maturation of CD4 and CD8αβ double negative, TCRαβ-expressing or TCRγδ-expressing T cells that directly migrate to the intestinal epithelium.80,81,82 The capacity of IELs to home to the intestinal epithelium is controlled by the expression of CCR9 on the surface of IEL precursors and its ligand CCL25.83,84 IELs also express the integrin αEβ7 (CD103) that can interact with E-cadherin on epithelial cells.85 Additionally, the mucosal environment leads to the induction of IELs from conventional TCRαβ+ cells that have matured as antigen-experienced cells in response to cognate antigen.75 In particular TGF-β, highly enriched in the gut, leads to the development of TCRαβ+CD8αα+ IELs and the coexpression of CD8α in conventional CD4+ T cells.86 IEL repetoire is polyclonal at birth and in response to exogenous stimuli, including luminal antigens and microbial composition, these cells expand to become oligoclonal in the adult.87 The development and expansion of IELs is also highly controlled by the cytokine IL-15 and by dietary metabolites. In particular, vitamin D and the arylhydrocarbon receptor, whose ligands are metabolized from the vegetable-derived phytochemical indile-3-carbinol, are required for the proper homeostasis of these cells.75,88,89 Although mice and human IELs share a number of features, some notable differences exist in terms of their composition and phenotype90 (see Table 34.1).
Intraepithelial Lymphocyte Functions
IELs have complex functions that include the promotion of local immunity concomitant with the maintenance of barrier integrity. Moreover, a role for IELs in promoting host defense has been revealed in various models of mucosal infection. For instance, TCRαβ+ CD8αβ+ IELs can promote effector responses to a variety of pathogens such as Eimeria vermiformis, Encephalitozoon cuniculi, and Toxoplasma gondii.91,92,93,94 In the context of bacterial or parasitic infections, IELs can mediate pathogen clearance via their capacity to produce various inflammatory mediators such as interferon (IFN)γ and TNF-α.95 IELs can also promote immunity to nematodes by releasing cytokines such as IL-13 that can promote mucus formation.96 Moreover, IELs express various natural killer (NK) receptors, endowing them with a strong cytolytic activity leading to the direct elimination of damaged or infected cells.75 Additionally, their protective role can result from the capacity of these cells to maintain the integrity of tight junctions between epithelial cells, therefore preventing epithelial transmigration of pathogens.97 In this regard, IELs have been associated with the maintenance of barrier function in the context of simian immunodeficiency virus (SIV) or human immunodeficiency virus (HIV).98,99 IELs can also exert regulatory function at mucosal sites. In particular, TCRγδ IELs play a major anti-inflammatory role via their capacity to promote tissue repair upon injury and to release an array of factors with regulatory properties. In particular, γδ T cells produce large amounts of keratinocyte growth factor, which plays an important role in supporting epithelial integrity.100,101 Further, these cells can abundantly produce the immunoregulatory cytokines IL-10 and TGF-β that can both limit inflammatory responses at mucosal sites.102,103,104 In mice, TCRγδ IELs express predominantly Vγ5 and Vγ1, whereas in humans TCRγδ IELs express essentially the Vγ1 gene segment.38 To date, little is known about the specificity of these cells.
Although the role of IELs is to maintain mucosal homeostasis, various lines of evidence suggest that in defined settings, IELs can also contribute to tissue damage and inflammation. For instance, the number of TCRγδ+ cells correlates with disease severity in inflammatory bowel disease (IBD) patients and, experimentally, these cells have been shown to promote mucosal inflammation.105,106,107,108 Further, CD8αβ+ IELs can also contribute to the initiation of experimental colitis. More particularly, IELs have been associated with the exacerbation of celiac disease, an inflammatory disorder in genetically susceptible individuals associated with the induction of inflammatory T-cell responses against dietary gluten.90 Celiac disease is characterized by a dramatic increase of the IEL compartment.90 TCR-activated CD8αβ+ TCRαβ+ IELs cause severe villous atrophy by targeting intestinal epithelial cells that express stress-induced major histocompatibility complex class I polypeptiderelated sequence antigen in an NKG2D-dependent manner.109,110 Additionally, IL-15, a factor that is overexpressed in various mucosal disorders including celiac and Crohn diseases,90 can trigger potent cytotoxic responses by CD8 αβ+ IELs via a NKG2D-DAP10-dependent signaling pathway.109 Supporting a role for IELs in celiac disease pathogenesis, genome-wide association studies have identified a number of genes involved in the development, migration, and cytotoxic/NK function of IELs.111
Mucosal Mononuclear Phagocytes
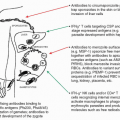
Immune reactivity against nonpathogenic gut elements is not only wasteful but is also known to lead to severe tissue damage. However, the development of active immunity is required to protect the host against invasive pathogens. This tight equilibrium is under the control of a complex network of mucosal mononuclear phagocytes (MPs) with unique properties (Fig. 34.1). Mucosal MPs form an organized cellular network in the LP and are distributed throughout the GALT.112 In the LP, major histocompatibility complex IIhi CD11chi MPs can be subdivided into two main populations defined as CD103+CD11b+CX3CR1- and CD103-CD11b+CX3CR1+. Although some debate exists in the
literature, the current consensus is that CD103+ cells are mucosal DCs with capacity to rapidly migrate into lymphoid structures, whereas the CX3CR1+ populations are considered to be tissue resident macrophages.113 The gut also contains a minor population of CD103+CD11b-CD8α+CX3CR1-DCs believed to be associated with ILFs.114 Additionally, the gut is home to plasmacytoid DCs.115 APC subset distribution varies anatomically with CD103+CD11b+ DCs mostly represented in the duodenum and CD103-CD11b+ DCs enriched in the large intestine.116
literature, the current consensus is that CD103+ cells are mucosal DCs with capacity to rapidly migrate into lymphoid structures, whereas the CX3CR1+ populations are considered to be tissue resident macrophages.113 The gut also contains a minor population of CD103+CD11b-CD8α+CX3CR1-DCs believed to be associated with ILFs.114 Additionally, the gut is home to plasmacytoid DCs.115 APC subset distribution varies anatomically with CD103+CD11b+ DCs mostly represented in the duodenum and CD103-CD11b+ DCs enriched in the large intestine.116
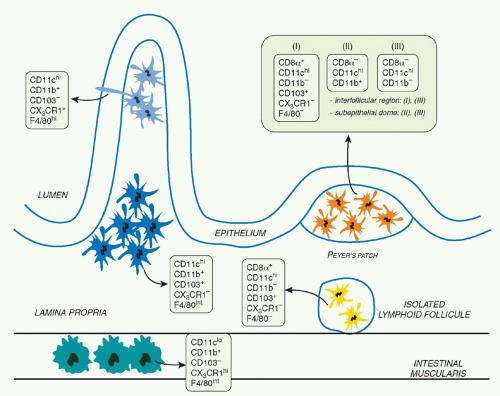 FIG. 34.1. Composition of the Gastrointestinal Mucosal Mononuclear Phagocyte Network. |
Origin of Mucosal Mononuclear Phagocytes
In the intestine, DC-restricted progenitors (termed common DC precursors) and pre-DCs give rise to CD103+CD11b+ and CD103+CD11b- populations.113,117 In contrast, CD103-CD11b+ cells are monocyte derived.113,117 Various cytokines, including FMS-like tyrosine kinase 3 ligand, granulocyte macrophage-colony stimulating factor, and macrophagecolony stimulating factor, control MP development and homeostasis.118 In the gut, these cytokines have a differential effect on the various MP subsets. Flt3L controls the differentiation of CD103+ DC (CD11b+ and CD11b-), granulocyte macrophage-colony stimulating factor the differentiation of CD103+CD11b+ DCs, and macrophage colony-stimulating factor receptor (M-CSFR) controls the homeostasis of CD103-CD11b+ cells.113,117,119 The fractalkine receptor CX3CR1 also controls the development of CD103-CD11b+ CX3CR1+ cells, likely via its capacity to control the recruitment of progenitors to the tissue.120 A number of transcription factors have been associated with DC lineage specification.121 In the gut, CD103+CD11b- DCs are highly dependent upon the transcription factors Id2, Irf8, and Batf3, whereas CD103+CD11b+ DCs are Notch2 dependent.113,122,123,124
Antigen Uptake and Cell Trafficking
Luminal antigens, including macromolecules, bacteria, and viruses, gain access to the cells of Peyer patches and ILFs through microfold cells, present in the follicle-associated epithelium.125,126,127 Microfold-cell transport is promiscuous and mediated by binding to surface-expressed carbohydrates in regions free of overlying mucus, but can be enhanced by the presence of antigen-specific IgA by immune targeting with anti-M-cell antibodies128,129 or by oral administration of toll-like receptor (TLR)2 or TLR4 ligands.130 A second site for antigen entry into the intestine is the nonfollicular absorptive epithelium, where both soluble
antigens and bacteria can gain access to DCs in the LP. This can occur by trans- or paracellular transport or by receptormediated trafficking, such as occurs through the neonatal FcR expressed on absorptive epithelial cells in humans.131 Luminal antigens can also be directly sampled by mucosal DCs. Indeed, subepithelial DCs can penetrate the epithelium monolayer by extending dendrites into the lumen, allowing them to sample particles and bacteria.132,133,134 In the terminal ileum, this process is tightly dependent on the fractalkine receptor CX3CR1 and controlled by the microbiota in an MyD88-independent manner.132,133,135 In addition to CX3CR1, CCL20 can also contribute to transepithelial dendrite formation.133 Luminal antigen can gain access to mucosal sites by direct damage to the epithelium, as can occur during IBD or in response to infection with HIV136 or Shigella flexneri.137 Antigen sampling may also occur by uptake of exosomes from epithelial cells or across villous microfold cells.138 Even in the absence of infection or inflammation, LP DCs constitutively traffic to MLN,139 which appears to be a relatively active process. These migratory DCs can carry self- or cell-associated antigens from apoptotic epithelial cells140 or soluble proteins given orally.141 Soluble antigens given orally can be processed by LP DCs, which then migrate to the MLN in a CCR7-dependent manner.142 Various lines of evidence support the idea that CD103+ DCs have enhanced migratory capacity to lymphoid structures compared to other mucosal MP subsets. Indeed, CD103+ DCs are highly represented in the gut afferent lymphatics,119 constitutively migrating to the MLN at steady state, and are the first to reach the MLN under inflammatory conditions.113,119 In response to inflammation, various signals including lipopolysaccharides and flagellin can influence the capacity of mucosal DCs to transport intestinal commensals and pathogenic bacteria to the MLN.143,144 A remarkable feature of this process is that commensal loaded LP DC migration is restricted to the mucosal immune compartment by the MLN.143 Such a process is believed to contribute to the compartmentalization of mucosal immune responses. Although as further discussed, mucosal MP can promote the induction of tolerance under steady-state conditions, the function of these cells is highly contextual, and under inflammatory settings mucosal MP can rapidly adopt a highly inflammatory phenotype and induce effector responses.145
antigens and bacteria can gain access to DCs in the LP. This can occur by trans- or paracellular transport or by receptormediated trafficking, such as occurs through the neonatal FcR expressed on absorptive epithelial cells in humans.131 Luminal antigens can also be directly sampled by mucosal DCs. Indeed, subepithelial DCs can penetrate the epithelium monolayer by extending dendrites into the lumen, allowing them to sample particles and bacteria.132,133,134 In the terminal ileum, this process is tightly dependent on the fractalkine receptor CX3CR1 and controlled by the microbiota in an MyD88-independent manner.132,133,135 In addition to CX3CR1, CCL20 can also contribute to transepithelial dendrite formation.133 Luminal antigen can gain access to mucosal sites by direct damage to the epithelium, as can occur during IBD or in response to infection with HIV136 or Shigella flexneri.137 Antigen sampling may also occur by uptake of exosomes from epithelial cells or across villous microfold cells.138 Even in the absence of infection or inflammation, LP DCs constitutively traffic to MLN,139 which appears to be a relatively active process. These migratory DCs can carry self- or cell-associated antigens from apoptotic epithelial cells140 or soluble proteins given orally.141 Soluble antigens given orally can be processed by LP DCs, which then migrate to the MLN in a CCR7-dependent manner.142 Various lines of evidence support the idea that CD103+ DCs have enhanced migratory capacity to lymphoid structures compared to other mucosal MP subsets. Indeed, CD103+ DCs are highly represented in the gut afferent lymphatics,119 constitutively migrating to the MLN at steady state, and are the first to reach the MLN under inflammatory conditions.113,119 In response to inflammation, various signals including lipopolysaccharides and flagellin can influence the capacity of mucosal DCs to transport intestinal commensals and pathogenic bacteria to the MLN.143,144 A remarkable feature of this process is that commensal loaded LP DC migration is restricted to the mucosal immune compartment by the MLN.143 Such a process is believed to contribute to the compartmentalization of mucosal immune responses. Although as further discussed, mucosal MP can promote the induction of tolerance under steady-state conditions, the function of these cells is highly contextual, and under inflammatory settings mucosal MP can rapidly adopt a highly inflammatory phenotype and induce effector responses.145
VITAMIN A AND MUCOSAL IMMUNITY
In the early 20th century, various studies identified dietary constituents that were essential for mammalian health. A single factor present in lipids was essential for growth and survival, which they termed “fat soluble factor A.”146 Subsequently designated vitamin A, studies over the years have demonstrated the pleiotropic influence of this nutrient, ranging from promoting eyesight and organogenesis to metabolism and immunologic fitness.147,148,149,150 In particular, the vitamin A metabolite RA plays a central role in the control of various aspects of innate and adaptive immune responses at mucosal sites.151,152 The effects of RA in mucosal immunity range from promoting effector cell migration to mucosal tissue and IgA generation, to the control of oral tolerance.153
Acquisition, Storage, and Metabolism of Vitamin A
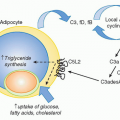
Vitamin A is a fat-soluble essential nutrient obtained from foods containing vitamin A precursors (ie, carotenoids) or vitamin A itself in the form of retinyl esters.154,155 Following absorption and arrival into circulation, retinyl esters enter the liver, where most of the vitamin A in the body is stored (Fig. 34.2).156 Liver retinyl esters are continually hydrolyzed into retinol and deployed into circulation.157 Once inside a cell, widely expressed alcohol dehydrogenases oxidize retinol into retinal, which can then bind to more selectively expressed retinaldehyde dehydrogenases (RALDHs) for oxidation into RA. Prior to its action, RA binds to nuclear receptors, including retinoic acid receptors (RARs) and retinoid X receptors.158,159 Notably, all-trans RA exclusively binds retinoid X receptors via heterodimers with the RAR family, which consists of three receptors: RAR alpha (RARα), beta (RARβ), and gamma (RARγ).158 Although RA is constitutively present in serum at low levels,160 RALDH expression is tightly controlled.147 GALT DCs express messenger ribonucleic acid for Aldh1a2, the gene encoding RALDH2.119,161,162 In particular, basal Aldh1a2 expression in GALT DCs is enriched in CD103+ DC subsets.162,163,164 Vitamin A itself is absolutely required for DC production of Aldh1a2 during homeostasis.162,165,166 Although dispensable at steady state, microbial stimuli may affect RALDH expression during an inflammatory or infectious response.167,168,169 In addition to DCs, several nonhematopoietic lineages within the GI tract and GALT, such as epithelia and stromal cells, share the capacity to synthesize RA (see Fig. 34.2).161,166,170,171,172
Role of Vitamin A in Migration to Mucosal Sites
Appropriate immune responses depend on the ability of effector and regulatory lymphocytes to home to the site of infection or injury. In this regard, DCs have been shown to foster lymphocyte migration into tissues where antigen was initially encountered.173 One of the dominant actions of vitamin A on the mucosal immune system is associated with the capacity of its metabolite RA to promote the migration of effector cells to mucosal tissues. In particular, CD103+ DCs from the GI tract and GALT can induce the mucosal homing markers—integrin heterodimer α4β7 and chemokine receptor CCR9—on stimulated effector T and B cells.163,174,175 The insight that mucosal DCs triggered α4β7 and CCR9 through their capacity to synthesize RA was based on the seminal observation that adding RA to T cells during activation selectively induced these gut homing markers.161 In a reciprocal fashion, blockade of RAR-mediated signaling and transcription in cultures containing GALT DCs reversed induction of α4β7 and CCR9.161,176 Recently, RA was revealed to predominantly affect the α4 subunit of α4β7 via binding of RARα to a retinoic acid receptor response element within the regulatory region of the α4 gene.177,178 An RA response element half-site was also recently discovered in the promoter region of CCR9, which RARα/retinoid X receptor heterodimers were able to bind to.179 These data together with the prominent expression of Rarα, the gene encoding RARα, in CD4+ T cells pinpoint RARα as a dominant mediator of CD4+ T-cell trafficking into mucosal sites.
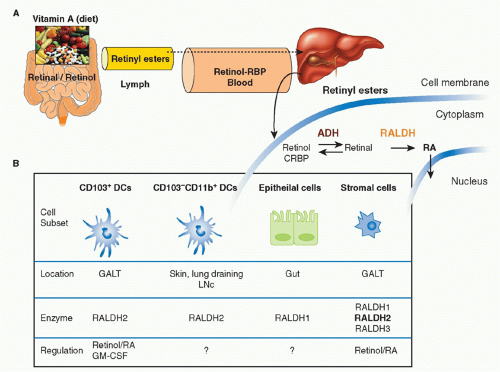 FIG. 34.2. Metabolism and Major Cellular Sources of Retinoic Acid (RA). Dietary vitamin A is absorbed in the intestine and transported through the lymphatics into the circulation where it enters the liver for storage. Retinol chaperoned by retinol binding protein is constitutively deployed from the liver into circulation. It is also secreted in bile that drains into the small intestine. Upon entry into cells, retinol is reversibly oxidized into retinal via the alcohol dehydrogenase enzyme family. Depending on the cell type (see Table 34.1), retinal can undergo irreversible metabolism into RA via retinal dehydrogenases (RALDH). Table 34.1 provides an overview of major cellular sources of RA at steady state including cellular location, the isoform(s) of RALDH that are expressed, and the factors known to induce RALDH expression in each cell type. |
Retinoic Acid in Infection and Immunity
Recent human data highlight the correlation between vitamin A status and T-cell function.180 Impaired and/or dysregulated T-cell responses have been observed in various models of infection and vaccination strategies in the context of vitamin A and retinoid receptor deficiency.181,182,183,184,185 Experimentally, these results support a role for RA in the development of both Th1- and Th17-cell responses (Fig. 34.3).181,182,183,184,185 RA signaling appears to control the fate of T-cell immunity largely through RARα and RARγ.184 In particular RA controls the capacity of CD4+ T cells to respond to antigen by regulating TCR signaling.184 Further, RARα regulates the maturation of DCs in the GALT165 and their ability to drive inflammatory responses in certain pathologic settings.186 These data illustrate the ability of RA to regulate a network of innate and adaptive immune cell functions that, through nonredundant receptor signaling pathways, support functional immune responses. The discovery that RA served as a critical factor in the generation of IgA-secreting B cells offered further evidence of a multifactorial role for RA in mucosal immunity.67 A number of studies have demonstrated the potent capacity of DCs from the intestinal LP, MLN, and Peyer patches to drive naïve B-cell differentiation into IgA+ B cells,68,187,188 and the ability of stromal derived cells to support IgA+ class switching in activated B cells.189,190 Synthesis of RA by GALT DCs was crucial for the generation of IgA by B cells, as antagonism of RA signaling significantly reduced IgA production.68,188 Complementing this finding, addition of RA to DC cocultures in which DCs lacked the capacity to synthesize RA restored IgA+ production. Notably, microbial induced cytokines, such as IL-6, were also integral cofactors in this process.68,188 This was also the case when stromal LP cells or stromal-derived follicular DCs from Peyer patches and MLN were assayed for their ability to foster the generation of IgA+ B cells.189,190 In a manner analogous to peripheral DCs, peripheral follicular DCs were able to efficiently support IgA+ production only when treated with RA and in the context of Myd88-dependent microbial stimulus. 190 Intriguingly, this gain of function was dependent on the ability of RA signaling to induce secretion of TGF-β in peripheral follicular DCs.191 RA signaling was shown to promote a similar effect (ie, induction of TGF-β production) in
bone marrow-derived DCs via inhibition of suppressor of cytokine signaling 3 activity.192 These findings suggest interdependency between TGF-β- and RA-propagated signals in several cell lineages.
bone marrow-derived DCs via inhibition of suppressor of cytokine signaling 3 activity.192 These findings suggest interdependency between TGF-β- and RA-propagated signals in several cell lineages.
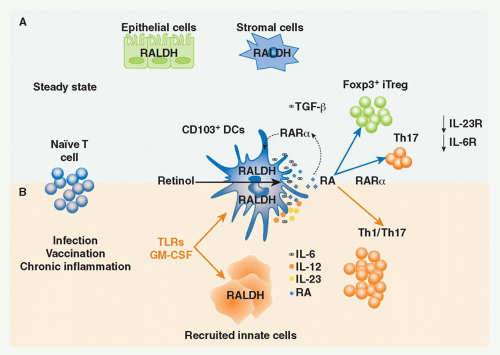 FIG. 34.3. Contextual Role of Retinoic Acid (RA) in Mucosal Immunity. Gut-associated lymphoid tissue is an RA-rich environment containing a large number of RA-synthesizing cells, including resident epithelia (intestinal epithelial cells) and stromal cells, as well as migratory cluster of differentiation (CD)103+ dendritic cells (DCs). Under steady-state conditions (top), RA sustains oral tolerance and helps maintain barrier integrity. These processes are mediated in large part by the ability of RA to support the induction of Foxp3+ inducible regulatory T and Th17 cells. Lamina propria CD103+ DCs induce and recruit heterogeneous CD4+ T-cell populations at steady state as a result of their ability to respond to commensal derived signals and produce both RA and transforming growth factor-β. During inflammation or infection (bottom), the inflammatory milieu triggers altered cytokine production by CD103+ DCs, leading to retinoic acid receptor α-dependent effector CD4+ T-cell activation and differentiation. Innate cell populations, including antigen-presenting cells, are recruited during inflammation and also contribute to RA production. Factors in the inflammatory milieu, including toll-like receptor ligands and the cytokine granulocyte macrophage-colony stimulating factor, promote retinaldehyde dehydrogenase activity in CD103+ DCs and potentially in recruited innate cells. |
Another significant source of IgA production is B1 B cells, which contribute to homeostatic mucosal integrity and early responses to pathogens. RA was recently shown to be required for the steady-state maintenance of this compartment through direct regulation of NFATc1.86 Combined with the capacity of RA to generate IgA+ B cells and facilitate their mucosal localization, vitamin A deficiency leads to severe decreases both in intestinal and serum IgA levels.86,188 Altogether, these findings underscore the importance of RA in IgA responses and humoral immunity.
Resulting from its pleiotropic effects on the mucosal immune system, RA can also, in a context-dependent manner, contribute to mucosal inflammation (see Fig. 34.3). Almost 30 years ago, mice fed a diet high in vitamin A were observed to exhibit more vigorous responses against grafts and tumors.193,194 These findings suggested that elevated retinoid levels could potentially predispose an individual to exuberant immune responses. In this regard, exposure to RA during inflammation contributed to loss of oral tolerance and reactivity to dietary glutens in a mouse model of celiac disease.186,195 More specifically, RA was demonstrated to synergize with IL-15 to promote mucosal DC production of IL-12p70, which exacerbated responses to the glutenderived antigen, gliadin.186 These data resonate with several reports, describing a possible association between pharmacologic retinoid treatment and spontaneous development of IBD, and point to vitamin A metabolic pathways as potential instigators of chronic inflammation.196,197
IMMUNE REGULATION
The mucosal environment requires a complex network of immunoregulatory mediators to maintain its integrity at steady state and in the face of infection or inflammation. Because of the extraordinary antigenic pressure at these sites, many mucosal cell types can in a context-dependent manner exert
regulatory activities. Under homeostatic conditions, the coordinated action of these cells results in the induction of regulatory responses toward dietary or commensal derived antigens.
regulatory activities. Under homeostatic conditions, the coordinated action of these cells results in the induction of regulatory responses toward dietary or commensal derived antigens.
Oral Tolerance
The acquisition of oral tolerance—the active suppression of inflammatory responses against the myriad of antigens derived from food and the microbiota—is a fundamental aspect of the mucosal immune system. Multiple mechanisms are involved in the induction of oral tolerance.198 Experimentally, oral administration of soluble proteins results in systemic tolerance that prevents the initiation of potentially damaging inflammatory responses upon rechallenge with the antigen in the periphery.199,200,201 A complex regulatory network, including specialized populations of APCs, lymphocytes, and innate cytokines, controls GI tract homeostasis and converges to favor the induction of regulatory responses toward antigens present at mucosal sites. Early reports suggested that commensals played a role in this process as oral tolerance could not be induced in the absence of gut flora or gut flora-derived signals.198,202,203,204,205 Over the last few years, various populations of Treg cells have been ascribed a central role in the induction of oral tolerance. CD4+ Treg cells, in particular, play an essential role in maintaining peripheral immune tolerance as well as preventing autoimmunity and chronic inflammation with several subsets described on the basis of their origin, generation, and mechanism of action. In a simplistic manner, these cells can be divided into endogenous Treg cells, represented by thymically derived Foxp3+ Treg cells and inducible Treg cells, such as IL-10-producing Treg cells206 or inducible Foxp3+ Treg (iTreg) cells.207,208 Although the precise mechanisms by which these cells coordinate their functions to maintain the delicate balance between immunity and tolerance remain incompletely understood, it can be conceived that collaboration and cross-talk among the aforementioned Treg cell populations is required for the integrated control of mucosal immune responses (Fig. 34.4).
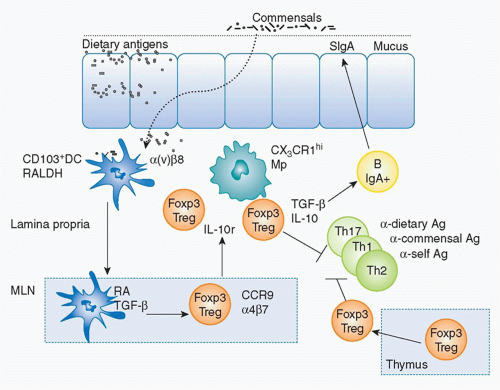 FIG. 34.4. Induction of Foxp3+ Regulatory T (Treg) Cells at Mucosal Sites. Under steady-state conditions, cluster of differentiation (CD)103+ dendritic cells (DCs) uptake directly or indirectly commensals and dietary antigens, and constitutively migrate to the mesenteric lymph node (MLN). In the MLN, based on their capacity to metabolize vitamin A (retinaldehyde dehydrogenase) and produce retinoic acid (RA) and to activate transforming growth factor (TGF)-β (α(v)β8+); CD103+ DCs induce Foxp3+ Treg cells from naïve T cells. Exposure to RA induces gut homing receptor CCR9 and α4β7 that allows the migration of these cells to the gut mucosa. In the gut, Foxp3+ Tregs can be expanded by CX3CR1 macrophages in an interleukin-10-dependent manner. At mucosal sites, inducible Tregs and thymically derived Tregs can limit a range of effector responses directed against dietary or commensal-derived antigens. Further, Treg cells in a TGF-β-dependent manner can also promote immunoglobulin A responses that in turn limit contact with the microbiota.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|