Fig. 6.1
Tissue processing and tumor characteristics involved in specimen adequacy assessment. Tissue processing fixatives and decalcification methods that are suitable for molecular testing are designated with a green check, and those that are contraindicated for use are designated with a red “X”. Tumor characteristics important in adequacy assessment include two-dimensional size, % tumor cellularity in the area sampled (%, tumor nuclear content/total cellular nuclear content including inflammatory and stromal cells), and the presence of absence of necrosis
The second and often more challenging factor to consider is the percent tumor cellularity present in the sample. When determining cellularity, it is important to remember that it is the nuclear content that should be assessed, not merely the area on the slide that the tumor cells are occupying. For example, inflammatory cells are much smaller than most tumor cells and can be present in a high density, which can contribute to a large percentage of the total DNA content present. Additionally, there are many instances in which a smaller specimen with a higher tumor cellularity (e.g., cytologic specimen) is preferable to a larger specimen with a very low tumor cellularity (Fig. 6.2). This is due to the fact that “contaminating” non-tumor DNA will dilute the mutant allele fraction, which could then fall below the analytic sensitivity and limit of detection present in the molecular testing platform. However, estimating the true tumor cellularity by eye can be difficult, and studies have shown poor interobserver variability [1].
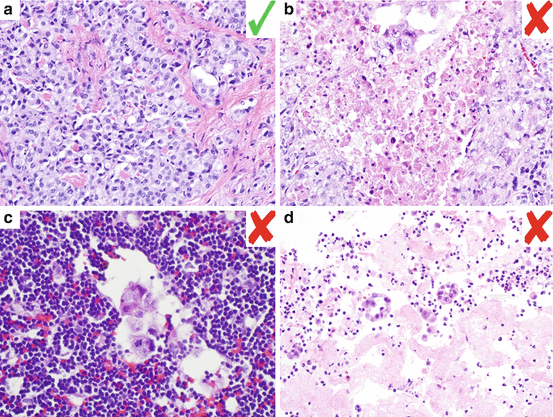
Fig. 6.2
Histologic examples of adequate and inadequate lung cancer specimens for molecular testing. (a) H&E histologic section from a lung adenocarcinoma specimen that is adequate for molecular testing with high tumor cellularity (minimal stromal and inflammatory cells) and no necrosis. (b) H&E histologic specimen from a lung adenocarcinoma specimen that is inadequate for molecular testing due to abundant necrosis. (c) H&E histologic section from a lymph node metastasis that is inadequate for molecular testing due to very low tumor cellularity. (d) H&E cytology cell block specimen that is inadequate for molecular testing due to scarcity of tumor cells and low overall tumor cellularity
Necrosis is often encountered in pathology specimens and can significantly alter the quality of the DNA isolated (Fig. 6.2), as necrotic cells undergo rapid nonspecific digestion of nucleic acids. However, the effect of necrosis on molecular testing is variable and depends on the percentage of the cells that are necrotic. While there are no strict guidelines dictating percent necrosis acceptable for molecular testing, studies across multiple tumor types have chosen <20% as the cutoff [2–4]. Thus, if possible, it is best to select specimens that have minimal tumor necrosis present or perform a tumor enrichment strategy such as microdissection to avoid necrotic regions. Although degraded DNA samples are not suitable for most molecular techniques, optimization strategies exist for DNA fragments <100 bp, many of which have been developed for use in forensic pathology. Such strategies include ligase detection coupled with PCR [5], as well as the use of short amplicons in multiplex PCR assays [6].
Finally, there are multiple factors related to sample handling that can significantly influence the suitability of the specimen for molecular testing (Fig. 6.1). The most commonly used fixative that has been shown to be well suited for the use in molecular testing is neutral buffered formalin [7, 8], which is the most widely used and preferred fixative for molecular testing [9]. In contrast, any fixatives that contain strong acids or heavy metals are generally not suitable for molecular diagnostics.
Importantly, the adequacy of a particular specimen must also be interpreted in the specific context of the molecular assay that is to be performed, as the requirements will vary based on each test.
Tissue Processing and Fixatives
The most commonly used fixative suited for use in molecular testing is neutral buffered formalin (NBF) . Several types of DNA damage have been identified in formalin-fixed tissues, which can serve as sources of sequence artifacts. Formaldehyde , the main component of formalin, is highly reactive with DNA bases and proteins and generates various cross-links between protein, DNA, and histones. Formaldehyde cross-links DNA by reacting with imino groups involved in base pair hydrogen bonds, thereby weakening the bonding strength of double-stranded DNA [10]. Additionally, formaldehyde cross-links DNA bases with histones, which results in a conformational change, also reducing the stability and causing partial denaturation of double-stranded DNA [11]. Deamination of cytosine bases is also seen in formalin-fixed tissues [12–14]. On average, molecular testing on formalin-fixed tissue requiring DNA segments less than 300 base pairs is successful, while those requiring longer stretches of DNA are less successful [15]. Formalin fixation also causes random nucleotide base substitutions, which can lead to false-positive results, and is particularly problematic in the setting of low DNA concentrations, in detection of rare variants, or in ultrasensitive assay designs [16–18].
In contrast, unbuffered formalin oxidizes into formic acid over time, which subsequently causes degradation of nucleic acids [19]. Similarly, any fixatives containing acids are also not suitable for molecular testing, including Bouin’s fixative, which contains picric and acetic acid, as acidic solutions fragment DNA significantly [20, 21].
When the specimen of interest is a bony metastasis, the decalcification process is a common problem for molecular laboratories. Most commercial formulations of decalcification solution contain acid, either mineral or organic acids, which are used to rapidly decalcify bone specimens. The best solution to decalcify tissues needed for molecular testing is to use non-acid-containing chelating solutions, such as EDTA, as it leaves the nucleic acids relatively intact [21, 22]. However, the slow rate of EDTA-based decalcification is generally incompatible with most surgical pathology workflows given expected turnaround times. Several commercial solutions and other supplemental methodologies are available to speed the process of decalcification when using EDTA, such as the addition of heat and agitation. Due to increasing requests for molecular testing on many specimen types, including core biopsies from bony metastases, there will likely be future demands to process bony specimens in such a way that preserves suitability for molecular testing.
Alcohol, in the form of 70% ethanol, is another fixative that is suitable for molecular testing, as it has been shown to preserve DNA as well as, or even superior to, neutral buffered formalin [23–25]. Alcohol is not a favored fixative for tissue specimens in routine histology laboratories for a multitude of reasons, including cost and compatibility with other FDA-approved ancillary studies; however, it is commonly used in cytology preparations. Therefore, many cytology specimens are indeed suitable for DNA-based molecular testing.
In addition to acid, any fixatives containing heavy metals (mercury, zinc, etc.), such as B plus, B5, and Zenker, will interfere with molecular testing. While the DNA itself may be intact, the heavy metals in these solutions compete with the cofactor magnesium that is required for most DNA polymerases and other enzymatic reactions [26–28].
Determining Assay Requirements
Selection of a molecular diagnostic testing platform depends on the clinical question that is being asked, the timing of test requests relative to clinical management, as well as the specimens that are available for testing. Additionally, the testing platform that is employed will influence what mutant allele fraction can be detected as well as how much DNA input will be required (Table 6.1). Before the widespread implementation of massively parallel sequencing, most of the molecular diagnostic assays available were single gene assays. While Sanger sequencing has long been considered a gold standard for sequencing methodologies, significant disadvantages to this technique include the limitation of analyzing a single amplicon (such as a single gene exon) at a time and low analytic sensitivity. As the analytic sensitivity of Sanger sequencing is approximately 25% mutant allele fraction, this requires a tumor specimen with at least 50% tumor cellularity, which would exclude a significant number of specimens from testing [29, 30].
Table 6.1
Molecular diagnostic testing platform comparison
Technique | Analytic sensitivity (% mutant allele) | DNA requirement | References |
|---|---|---|---|
Sanger sequencing | 15–20% | 100 ng | |
PCR-RFLP | 1–5% | 50 ng | |
Real-time PCR | 0.5–5% | 15–20 ng | |
Digital PCR | 0.1% | 10–50 ng | [40] |
Pyrosequencing | 5–10% | 25–100 ng | |
Single nucleotide base extension and size separation | 5–12.5% | 200 ng | |
Massively parallel sequencing | |||
Hybrid capture | 3–5% | 50–500 ng | [31] |
Amplicon | 3–5% | 10–50 ng | [32] |
Currently, in the realm of less expensive sequencing technology, multi-gene massively parallel sequencing assays are becoming more commonly used as a first-line test choice. While the large multi-gene panels are more comprehensive in their approach, they may not be as sensitive as other more targeted approaches, such as specific PCR-based assays. For example, if one is interested in identifying a subclonal event, such as a resistance mutation present in a small fraction of cells, a more tailored assay is better suited to answer that question. While both PCR- and NGS-based assays have their strengths and weaknesses, they can also be tailored to address specific needs and requirements.
PCR is a technique used to amplify a few copies of target DNA to several orders of magnitude, generating thousands to millions of copies of a particular sequence. Multiple PCR-based testing methods have been used routinely in molecular diagnostics, including quantitative PCR (real-time or QPCR ), allele-specific PCR , and digital PCR (dPCR) . PCR-based assays provide highly sensitive mutation detection (as little as 0.5% allele fraction), but only a single target can be interrogated in a single reaction and that target must be known a priori.
In contrast, massively parallel sequencing examines many different targets at once, with analytic sensitivity depending of assay design including breadth of coverage and depth of sequencing, but optimally down to 3–5% mutant allele frequency. Several different methodologies exist for library preparation prior to massively parallel sequencing, including hybrid capture and amplicon-based strategies. Hybrid capture approaches require a higher input DNA (~50–500 ng DNA) and use biotinylated probes that hybridize to the genes or exons of interest, which are subsequently purified, modified with adapters, and then sequenced [31]. In contrast, amplicon-based methods utilize PCR primers to the genes and exons of interest with multiple subsequent PCR amplification rounds, thus requiring less input DNA (~10 ng DNA) [32].
While the basic limitations of each assay are as stated above, assays can be tailored to a specific target and question. For example, new assay designs incorporating molecular barcodes allow for the removal of significant noise occurring secondary to sequencing errors and can increase the analytic sensitivity of the assay to 0.1% mutant allele fraction [33]. Particular applications for which this would be helpful include serial disease monitoring for the emergence of a recurrent or resistant clone, particularly in the form of circulating tumor DNA (ctDNA) . As with any laboratory developed test, the lower limits of acceptability, with regard to sensitivity and specificity, must be determined for each assay that is developed.
DNA Yield from Archival Tissue Samples
As the majority of lung cancer specimens available for molecular diagnostic testing are formalin-fixed paraffin-embedded tissues (FFPE), the validation and use of these specimens in a variety of molecular testing platforms are central to the clinical implementation of genomic testing. Depending on the specific parameters of the specimen as detailed earlier in the chapter, the minimum tissue needed will vary based on testing platform. It has been extensively documented that multiple testing methodologies can be successfully performed on DNA isolated from archival FFPE tissues. For example, OncoPanel testing performed at the Center for Advanced Molecular Diagnostics at Brigham and Women’s Hospital had success with 96% of nearly 3900 archival FFPE specimens that were run on a targeted panel massively parallel sequencing assay with a minimum input of 50 ng DNA [47]. These success rates were obtained with minimum tissue requirements of at least 20% malignant cells and at least 3 mm in greatest linear dimension of tumor [47].
Archival cytology direct smears have also been shown to be suitable for molecular testing, as adequate DNA was successfully isolated for PCR with amplicons ~200 bp in length from direct smears that were at least 1 year old [48]. Direct smears stained with both Giemsa and Diff-Quik were shown to have DNA quality equal to those from matched frozen controls and were suitable for multitargeted sequencing on a large panel of genes [49]. In contrast to direct smears, increased DNA degradation over time has been documented in liquid-based cytology (LBC) specimens, as successful PCR amplification from archival LBC specimens required primers for PCR products <200 bp [48]. It is important to note that every assay must be validated for every specimen type to be used in the clinical diagnostic test.
Tumor Enrichment Techniques
Multiple techniques exist to isolate tumor cells from FFPE tissue blocks (Table 6.2). These techniques range from simple scrolls with no attempt to enrich for tumor content to more sophisticated methodologies that can capture even a single tumor cell. While it depends on the particular specimen, in most instances, it is necessary to perform some level of enrichment for tumor cells when isolating tumor DNA from FFPE blocks. This greatly increases the number of specimens that is adequate for molecular testing and enhances the suitability of the isolated input DNA for the particular analytic sensitivity of the molecular methodology in use. However, it is also important to keep in mind that the minimum tumor cellularity requirements are based on the assumption of no tumor heterogeneity, and subclonal events may be below the analytic sensitivity.
Table 6.2
Tumor enrichment techniques
Technique | Method | Technicality | Precision |
|---|---|---|---|
No enrichment | Thick micron scrolls, scraping entirety of unstained slide | ✓ | ✓ |
Macrodissection | H&E examined and marked by pathologist for subsequent tumor dissection without microscope | ✓✓ | ✓✓ |
Manual microdissection | H&E examined and marked by pathologist for subsequent tumor dissection under light microscope | ✓✓✓ | ✓✓✓ |
Laser capture microdissection | A laser-coupled microscope is used to capture the tissue of interest | ✓✓✓✓ | ✓✓✓✓ |
If no tumor enrichment technique is needed, for example, in tissue blocks which consist predominantly of tumor with minimal stroma or inflammatory cells, then either using thick (e.g., 50 micron) scrolls or scraping the entirety of a series of unstained slides would be adequate. Presuming the adequacy of the slide had been previously documented, these techniques would require no additional pathology review for subsequent tumor and DNA isolation. However, it is important to remember that even tissues, which appear to be >90% tumor by light microscopy, may be significantly contaminated by inflammatory and stromal cells. Additionally, tumor content can change as deeper sections into the block are obtained.
A common tumor enrichment technique that is employed by many molecular diagnostic laboratories is that of macrodissection , as it provides an adequate amount of tumor enrichment, yet requires limited technical expertise. In this procedure, a hematoxylin and eosin (H&E)-stained slide obtained from a tissue level immediately adjacent to a series of unstained slides is examined by a pathologist, and the area of interest is circled with a marking pen by the reviewer. This technique serves to remove large areas of tissue that do not contain tumor, as well as to concentrate the isolation on viable areas of the tumor that are free of necrosis and significant inflammation. The technologist who is isolating the tumor cells will use the marked H&E as a guide to scrape the appropriate area of the unstained slide or core the appropriate area of the block. Similarly, manual microdissection can be performed, in which the technologist isolating the tumor cells uses the aid of a light microscope when performing the dissection. This latter technique may be facilitated by the use of a stereomicroscope [50].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


