The Interleukin-1 Family
Charles A. Dinarello
Mihai G. Netea
INTRODUCTION
More than any other cytokine family, the interleukin (IL)-1 family of ligands and receptors is primarily associated with acute and chronic inflammation. Also, more than any other cytokine family, the IL-1 family plays a fundamental role in the nonspecific innate response to infection that facilitates specific immunologic responses such as antibodies and cytotoxic T-lymphocytes. This nonspecific response to infection is now termed the “innate immune response.” The cytosolic segment of each member of the IL-1 receptor family contains the toll-IL-1-receptor (TIR) domain. This domain is also present in each toll-like receptor (TLR), receptors that respond to microbial products, viruses, and nucleic acids. TIR is the functional domain for both the TLR and IL-1 receptor families, as mutations in this domain result in loss of response to IL-1 and TLR agonists. The biologic properties of both IL-1 family ligands and TLR agonists characteristically are proinflammatory and act as adjuvants for specific immune responses to antigen. Thus, the IL-1 family of ligands and receptors is fundamental to innate immunity. Of the 11 members of the IL-1 family, IL-1β has emerged as a therapeutic target for an expanding number of systemic and local inflammatory conditions termed “autoinflammatory” diseases. These diseases are distinct from autoimmune diseases and include rare hereditary conditions. But autoinflammatory diseases are also common diseases such as heart failure, gouty arthritis, and type 2 diabetes. For these, neutralization of IL-1β results in a rapid and sustained reduction in disease severity. Another member of the IL-1 family, IL-1α, is also a mediator of inflammation but is classified as an “alarmin” because the cytokine is present in most cells and readily released upon cell death. Whereas treatment for autoimmune diseases often includes immunosuppressive drugs, neutralization of IL-1β or blocking the IL-1 receptor is mostly anti-inflammatory.
With one exception, all members of the IL-1 family are initially translated as precursors lacking a signal peptide for secretion via the Golgi. The precursors are found in the cytosol and exit the cell following death by necrosis, not apoptosis. For example, once released, IL-1α, IL-33, and IL-36 can be processed extracellularly by neutrophil proteases into active cytokines. Although IL-1β is primarily processed intracellularly by the cysteine protease caspase-1, the IL-1β precursor can also be cleaved extracellularly into an active cytokine by similar serine proteases of neutrophils. The one member of the IL-1 family that is readily secreted is the IL-1 receptor antagonist (IL-1Ra). IL-1Ra is translated with a signal peptide (Fig. 26.1), although an intracellular form also exists.1 IL-1Ra is produced in health and is found circulating in mice and humans where the antagonist serves as a brake on inflammation driven by endogenous IL-1α or IL-1β. IL-1Ra binds to the IL-1RI and blocks the receptor from binding to either IL-1α or IL-1β (see Fig. 26.1C). Mice as well as humans born with a deficiency in functional IL-1Ra exhibit increased systemic and local inflammation; in humans, a deficiency in IL-1Ra is lethal. The IL-36 receptor antagonist (IL-36Ra), another member of the IL-1 family, inhibits the activity of endogenous IL-36α, β, and γ. Although IL-36Ra is not readily secreted, individuals with a mutation in IL-36Ra develop a severe form of psoriasis. One may conclude that most members of the IL-1 family primarily promote inflammation and enhance specific acquired immune responses. But there are also members that provide a brake on inflammation. The primary characteristics of the each member of the IL-1 family are depicted in Table 26.1.
INTERLEUKIN-1 FAMILY AND INNATE RESPONSES
Independent of the type of organism or its products, the innate response is one of inflammation in which the host musters its defenses to increase the production and infiltration of phagocytic cells to the area of the invading microbe in an attempt limit infection and kill-off the invader. Systemically, the liver increases the synthesis of acute phase proteins, include antiproteases. Even in humans, in most cases this process protects the subject without the use of antibiotics. For example, a break in the skin allows bacteria to gain access to the dermis and subsequent inflammation provides activation of complement, the release of preformed cytokines from keratinocytes, an increase in vascular wall adhesions molecules, and the extravasation of neutrophils. This response has functioned to battle against invaders for millions of years and can be traced back to fruit flies.
The skin, lung, and intestinal tract each provide a first line of defense against microbial invasion and the lining cells, whether keratinocytes of the skin, the alveolar epithelial cells of the pulmonary tree, or the epithelial cells of the entire gastrointestinal tract, each contain preformed IL-1α, IL-18, and IL-33 as well as the members of the IL-36 subfamily. Because these members of the IL-1 family are each preformed in these cells, their release is a consequence of injury and is immediate. Therefore, they are termed “alarmins” as they alert the host to initiate the response. There are other “alarmins” from the lining cells that participate in defense, for example, defensins, which are directly antimicrobial.
Each of the constitutively present IL-1 family members in lining cells is present as a precursor. In the case of IL-1α, the precursor is fully active; in the case of the other members, the precursors are weakly active at first but are converted to more active cytokines upon the infiltration of neutrophils and processing by extracellular neutrophil proteases. In the end, the infection is contained, the invading microorganism is eliminated, and skin begins its process of repair.
Each of the constitutively present IL-1 family members in lining cells is present as a precursor. In the case of IL-1α, the precursor is fully active; in the case of the other members, the precursors are weakly active at first but are converted to more active cytokines upon the infiltration of neutrophils and processing by extracellular neutrophil proteases. In the end, the infection is contained, the invading microorganism is eliminated, and skin begins its process of repair.
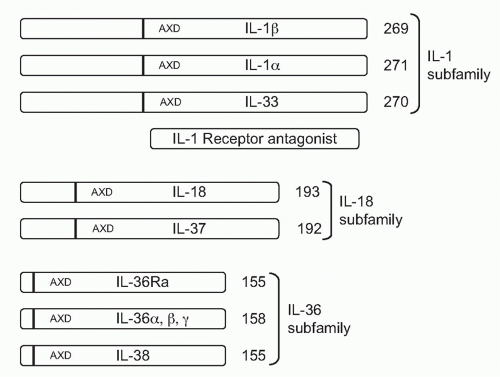 FIG. 26.1. Organization of the Interleukin (IL)-1 Family into Three Subfamilies. The number of amino acids of the fulllength of each member is shown at the C-terminal end. The consensus sequence (A-X-D) is common to all IL-1 family members and serves to locate the N-terminus nine amino acids forward from this site, as shown by the dark vertical bar. The N-terminus results in propieces of various lengths. The IL-1Ra has a bonafide signal peptide and is shown by comparison. |
Following the cloning of the mouse IL-1 receptor,2 the cytosolic domain of the IL-1 receptor was found to be homologous to toll of the fruit fly.3 Moreover, at the same time, the TIR domain for IL-1 signaling (Fig. 26.2A) was shown by Heguy to be required for IL-1 signaling.4 Toll had been initially studied since its discovery in 1985 because of its central role in establishing dorsal ventral polarity in Drosophila. Only since 1996 was toll linked to survival in fruit flies infected with fungi.5 However, it had already been reported, back in 1988, that a member of the IL-1/TLR family, human IL-1β, protected mice from lethal Pseudomonas infection.6 As noted previously, the TIR domain is essential for both IL-1 receptor family and TLR family signaling; a mutation in the TIR domain severely impairs responses to IL-1 family ligands as well to a large number of microbial products.7
TABLE 26.1 Interleukin-1 Family Members | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
The TIR domain binds MyD88 (see Fig. 26.2A and E), itself a TIR domain-containing protein, through TIR/TIR interactions triggering a cascade of kinases that propagate the IL-1 signal and result in transcription of a large number of genes, the majority of which code for other cytokines, chemokines, and a host of inflammatory mediators. Of these is IL-1 and other members of the IL-1 family such as IL-36 and IL-18.
The “innate immune response” regulates to the “acquired immune response.” The late Charles Janeway proposed that the innate response assists the host in mounting an acquired immune response. This relationship between a nonspecific cytokine providing help for a specific response to a microbial antigen is simply the adjuvant property of some cytokines. The adjuvant property of some cytokines functions by upregulating lymphocyte growth factors such as IL-2, IL-4, and IL-6, or lymphocyte receptors resulting in expansion of lymphocyte clones, which will either rid the host of the invading microorganism with neutralizing antibodies or in generation of cytotoxic T cells to eliminate viral infections. In 1979, purified human IL-1β, a nonspecific macrophage product, was shown to augment the T-cell response to specific antigen.8 It was nearly 20 years later that TLR were identified as inducing IL-1β from monocytes.
ORGANIZATION OF THE INTERLEUKIN-1 FAMILY OF LIGANDS AND THE CONSENSUS SEQUENCE
As depicted in Figure 26.1, the IL-1 family can be divided into subfamilies according to the length of the precursor and the length of the propiece for each precursor. The IL-1 subfamily is comprised of IL-1α, IL-1β, and IL-33. This subfamily has the longest proteins with the longest propieces. In the case of IL-1β, the propiece is cleaved intracellularly by caspase-1 and then the mature cytokine is secreted. In the case of IL-1α, cleavage appears to be by the membrane protease calpain, but
extracellular neutrophil proteases can also cleave the IL-1α precursor. Extracellular neutrophil proteases account for the cleavage of the propiece of IL-33. The exception in the IL-1 subfamily is IL-1Ra, which contains a signal peptide.
extracellular neutrophil proteases can also cleave the IL-1α precursor. Extracellular neutrophil proteases account for the cleavage of the propiece of IL-33. The exception in the IL-1 subfamily is IL-1Ra, which contains a signal peptide.
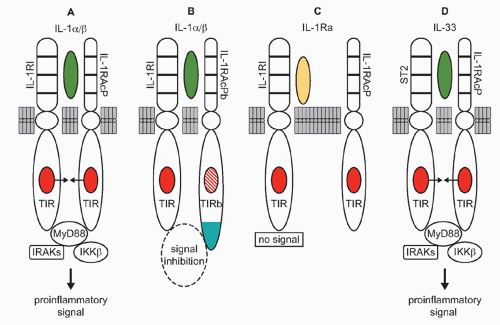 FIG. 26.2. Interleukin (IL)-1 Subfamily. A: IL-1α or IL-1β binds to the IL-1RI and recruits the coreceptor IL-1RAcP. The heterodimeric IL-1 receptor complex results in a close approximation of the toll-IL-1-receptor (TIR) domains on each receptor chain (arrows), resulting in the binding of intracellular MyD88 to the complex followed by phosphorylation of MyD88. Subsequent phosphorylations of IRAKs and IKKβ, increased NF-κB, and IL-1R AcP-1 translocation to the nucleus take place followed by expression of proinflammatory genes. B: In the central nervous system, IL-1α or IL-1β bind IL-1RI recruiting IL-1RAcP but can also recruit the coreceptor IL-1RAcPb. IL-1RAcPb contains an altered TIR domain, which results in a reduced signal. C: IL-1Ra binds to IL-1RI. There is no recruitment of the co-receptor IL-1RAcP, no approximation of the TIR domains, and no signal. D: IL-33 binds to its specific receptor, ST2, and recruits the coreceptor IL-1RAcP, the TIR domains approximate, and signal transduction is initiated resulting in the induction of proinflammatory gene profile. |
The IL-18 subfamily is comprised of IL-18 and IL-37. By comparison, this subfamily has a smaller propiece. IL-18 requires the cleavage of its propiece by caspase-1 in order to be active. IL-37 is part of the IL-18 subfamily because the cytokine binds to the IL-18Rα chain. It is unclear how the propiece of IL-37 is removed. The IL-36 subfamily comprised of IL-36α, β, and γ, as well as IL-36Ra. In addition, IL-38 likely belongs to this family due to its binding to the IL-36R. The IL-36 subfamily has the shortest propiece.
A consensus sequence in all members of the IL-1 family is A-X-D, where A is an aliphatic amino acid such as isoleucine, methionine, or leucine; X is any amino acid; and D is aspartic acid. The aspartic acid of the consensus sequence is not the aspartic acid of the caspase-1 cleavage recognition site. The A-X-D motif is conserved in the IL-1 family where it plays a role in three-dimensional structure of the active cytokine. The actual N-terminus is often located nine amino acids before the A-X-D site. By eliminating the amino acids before the N-terminus, the first beta-sheet structure common to all members of the IL-1 family can form. For example, with the tenth amino acid before A-X-D consensus site as the N-terminus, the specific activity of the IL-36 subfamily (IL-36α, IL-36β, IL-36γ and the IL-36Ra) is low. However, with the ninth amino acid as the N-terminus, there was a marked increased in the activity.9 In the case of IL-1β, the ninth amino acid before the A-X-D site coincides exactly with the N-terminal alanine generated by the caspase-1 site.
THE INFLUENCE OF INTERLEUKIN-1 FAMILY ON TH17 RESPONSES
The IL-1 family plays a significant role in interferon (IFN)γ production, which is essential for the defense against intracellular pathogens. On the other hand, Th2 cells are characterized by the production of IL-4 and are important in the host defense against parasitic infections. For more than one decade, the dichotomy between Th1 and Th2 has been the focus of studies on differentiation of cluster of differentiation (CD)4+ T-lymphocytes. More recently, Th17 helper cells have been described and are characterized by their production of IL-17. IL-17 plays a major role in neutrophil recruitment and host defense against extracellular bacteria and fungi. Th17 cells produce a distinct cytokine profile, namely IL-17A, IL-17F, IL-21, and IL-22. The cytokines produced
by Th17 cells, in addition to activating neutrophils, are also crucial for nonimmune cells, for example, induction of defensins by IL-22 in epithelial cells and keratinocytes, which are part of mucosal and skin defenses. It has become apparent that Th17 responses are associated with chronic inflammation and autoimmune diseases such as multiple sclerosis, type 1 diabetes, Crohn disease, and psoriasis. Furthermore, Th17 responses are fundamental for host defense against many microorganisms, although they also contribute to the inflammation during infection.
by Th17 cells, in addition to activating neutrophils, are also crucial for nonimmune cells, for example, induction of defensins by IL-22 in epithelial cells and keratinocytes, which are part of mucosal and skin defenses. It has become apparent that Th17 responses are associated with chronic inflammation and autoimmune diseases such as multiple sclerosis, type 1 diabetes, Crohn disease, and psoriasis. Furthermore, Th17 responses are fundamental for host defense against many microorganisms, although they also contribute to the inflammation during infection.
Whereas IL-4 and IL-12 were the first cytokines described as influencing Th-cell differentiation, cytokines of the IL-1 family also influence cytokine differentiation. IL-18 was initially described as IFNγ-inducing factor due to its strong stimulatory effect on Th1/IFNγ responses.10 It is now known that IL-18 is, in fact, a crucial cytokine directing the development of Th1 cells, and one role of IL-12 is the induction of the expression of IL-18 receptors. In contrast, binding of IL-33, another member of the IL-1 family, to its ST2 receptor plays a role in inducing Th2 responses,11 and it thus appeared as if distinct members of the IL-1 family of cytokines directed Th1 versus Th2 differentiation. Considering these effects of IL-18 and IL-33, it came as no surprise that IL-1, the most well-known member of the family, participates in the function of Th cells.
Known for over 30 years that IL-1 enhances T-cell activation and recognition of antigen, one of the early names of IL-1 was lymphocyte activation factor. The specificity of this response was, however, not known. Although initially only IL-23, IL-6, IL-21, and transforming growth factor-β were suggested to play a role in the development of Th17 responses in mice, there is no dearth of data that a more complex picture exists. Thus, IL-1β, IL-6, and transforming growth factor-β have been reported to induce the development of Th17 cells, whereas IL-23 has been reported to be important for the maintenance of Th17 cells. The combination of IL-23 and IL-1β induce the development of human Th17 cells expressing IL-17A, IL-17F, IL-22, IL-26, the chemokine CCL20, and transcription factor RORγt.12 Interestingly, these cells also released IFNγ, displaying a phenotype common to both Th17 and Th1 cells.12 The strong capacity of IL-1 to induce Th17 differentiation has been also linked to its well-known capacity to induce the release of prostaglandins, as reviewed in Dinarello.13 PGE2 induced by COX-2 is a stimulator of Th17 induction, and inhibitors of cyclooxygenase decrease IL-17 production.14 On the other hand, engagement of the aryl hydrocarbon receptor, a pathway demonstrated to be crucial for the generation of Th17 cells, has been shown to strongly induce IL-1β.15 In addition to inducing IL-17 production from the Th17 subset of lymphocytes, IL-1β is required for the production of IL-17 by natural killer (NK) T cells16 and of IL-22 from NK cells.17
Thus, cytokines of the IL-1 family have an important role in the differentiation of the Th subsets, with IL-1β strongly inducing Th17 responses, IL-18 being crucial for the generation of Th1 cells, and IL-33 being important in Th2 responses. Interestingly, reciprocal regulation has been demonstrated between the various Th subsets, with cytokines released by Th2 cells inhibiting Th1 responses, whereas IFNγ release from Th1 cells impairing both Th2 and Th17 responses.
INTERLEUKIN-1α
From an evolutionary viewpoint, IL-1α is the oldest member of the IL-1 family and its primary amino acid sequence is closely related to that of the fibroblast growth factor family. Like fibroblast growth factor, IL-1α does not have signal peptide, binds to nuclear deoxyribonucleic acid (DNA), exits the cell upon death, and binds to its receptor as an unprocessed precursor. As shown in Figure 26.2A, IL-1α binds to the IL-1RI and recruits the IL-1R accessory protein (IL-1RIAcP) to form a heterodimeric complex, which signals to induce inflammation. In health, primary cells contain constitutive levels of the IL-1α precursor but not IL-1β.18 The IL-1α precursor is present in keratinocytes, thymic epithelium, hepatocytes, endothelial cells, the epithelial cells of mucus membranes, including the entire gastrointestinal tract, and fibroblasts, regardless of their location. The propiece of IL-1α precursor can be cleaved extracellularly by neutrophil proteases, a step that increases its biologic activity. However, IL-1α can also be active as a membrane-associated cytokine. Most cell lines, including tumor cell lines, contain constitutive levels of IL-1α.19,20,21 Using an epithelial cell line, what was considered to be intrinsic IFNγ activities depended largely on constitutively expressed IL-1α. IFNγ activities were inhibited by antibodies to IL-1α, but not to IL-1β.20 The concept that IL-1α acts as an autocrine growth factor assumes that the intracellular IL-1α precursor regulates normal cellular differentiation, particularly in epithelial and ectodermal cells. In support of the concept, an antisense oligonucleotide to IL-1α reduces senescence in endothelial cells.22 In fibroblasts, the constitutive IL-1α precursor binds to HAX-1, a nonreceptor substrate for tyrosine kinases in hematopoietic cells. In fibroblasts, the IL-1α HAX-1 complex translocates to the nucleus.23 Although the concept is that IL-1α acts as an autocrine growth factor in fibroblasts or endothelial cells in vitro, the data should be interpreted carefully as mice deficient in IL-1α show no demonstrable defects in growth and development, including skin, fur, epithelium, and gastrointestinal function.24 However, mice deficient in IL-1α still retain the N-terminal propiece, which functions as nuclear factor.21 In fact, in another study, the N-terminal propiece of IL-1α was shown to bind HAX-1.25
Is there is a role for intracellular precursor IL-1α in normal cell function? The IL-1α precursor is present in cells that also contain large amounts of the intracellular form of the IL-1Ra (icIL-1Ra), as reviewed in Arend.1 This form of the IL-1Ra also binds to the IL-1 receptor and prevents signal transduction. In fact, icIL-1Ra is thought to compete with the intracellular pool of precursor IL-1α for nuclear binding sites.
Membrane-Associated Interleukin-1α
Precursor IL-1α can be found on the surface of several cells, particularly on monocytes and B-lymphocytes, where it is referred to as membrane IL-1α.26 Membrane IL-1α is biologically active27; its biologic activities are neutralized by antibodies to IL-1α but to IL-1β. Endothelial cells undergoing stress-induced apoptosis release membrane apoptotic bodylike particles containing nuclear fragments and histones as well as full-length IL-1α precursor and the processed mature
form.28 When injected into mice, apoptotic body-like particles containing the IL-1α precursor induce neutrophilic infiltration that was prevented by neutralization of IL-1α but not IL-1β.28
form.28 When injected into mice, apoptotic body-like particles containing the IL-1α precursor induce neutrophilic infiltration that was prevented by neutralization of IL-1α but not IL-1β.28
Processing and Secretion of Interleukin-1α
Although the IL-1α precursor is biologically active, the processed form is more active. Furthermore, the binding of IL-1α to the IL-1RI has been modeled using recombinant IL-1α with an N-terminus at 113. The processing of the IL-1α precursor is accomplished by calpain II, a membrane-associated, calcium-dependent cysteine protease.29 In macrophages treated with hydroquinone, calpain II levels fall and are associated with inhibition of IL-1α precursor processing.29 Not surprisingly, calcium influx induced IL-1α secretion of the processed form.30 The secretion of IL-1α requires the presence of IL-1β, as IL-1β-deficient mice do not secrete IL-1α.31 IL-1α binding to IL-1β has been reported in which IL-1β acts as a chaperone for the secretion mechanism via caspase-1.31 In another study, IL-1β was shown to bind to, and enhance the activity of, high-mobility group protein B1 (HMGB1).32 It is thus possible that both IL-1α exits the cell bound to IL-1β and HMGB1.
Biologic Functions of Constitutive Interleukin-1α: Interleukin-1α and Sterile Inflammation
Large numbers of reports use bacterial and fungal products to induce cytokines as models of inflammatory disease; however, most inflammatory diseases are sterile. For example, the inflammation associated with atherosclerosis, myocardial infarction, stroke, cancer, and renal and liver failure is sterile. The hypoxic insult that takes place in ischemia results in local necrosis and release of cellular contents, including nucleic acids. Members of the IL-1 family contribute to sterile inflammation, and IL-1α plays a significant role in this regard. Upon cell death by necrosis, the IL-1α precursor is released33,34 and binds to the IL-1 receptor on nearby tissue macrophages and epithelial cells, triggering a response.35,36 For example, infiltration of neutrophils occurs first and followed by influx of monocytes.35 Extracts of tumor cells induce neutrophilic inflammation, which does not occur in mice deficient in IL-1RI and is prevented by neutralization of IL-1α, not neutralization of IL-1β.37 Sterile inflammation is independent on TLR2 and TLR4.37
Thus, IL-1α, either the unprocessed precursor or the calpain cleavage form, is classified as an “alarmin” because the cytokine is preformed and triggers an inflammatory response rapidly. Endothelial cells subjected to nutritional stress release inflammatory apoptotic bodies, which contain both the precursor and processed forms of IL-1α.28 Inflammatory apoptotic bodies induce chemokine and neutrophilic infiltration into the peritoneal cavity, both of which are IL-1α dependent.28 Platelets also contain IL-1α as well as IL-1β.38 Platelet-derived IL-1 induces chemokines such as IL-8 from endothelial cells39 and monocyte chemotactic protein (MCP-1) from monocytes.40 Platelet-derived IL-1α is important in brain injury in stroke models41 and in atherosclerosis.42
Studies in Interleukin-1α-Deficient Mice
Mice deficient in IL-1α are born healthy and develop normally. In some models of local inflammatory responses, wild-type and IL-1α-deficient mice develop fever and acutephase proteins, whereas IL-1B-deficient mice do not.24 In addition, although the inflammation-associated induction of glucocorticoids was suppressed in IL-1β-deficient mice, this suppression was not observed in IL-1α-deficient mice. However, expression of IL-1β messenger ribonucleic acid (mRNA) in the brain decreased 1.5-fold in IL-1α-deficient mice, whereas expression of IL-1α mRNA decreased more than 30-fold in IL-1β-deficient mice. These data suggest that IL-1β exerts greater control over production of IL-1α than does IL-1α over the production of IL-1β. In caspase-1-deficient mice, IL-1α production is also reduced,43 further suggesting that production of IL-1α is under the control of IL-1β. It is important that caspase-1-deficient mice are also deficient in caspase-11.44
In mice fed a high-fat diet, serum amyloid A protein, a marker of inflammation in atherogenesis, was markedly lower in IL-1α-deficient mice compared to wild-type or IL-1β-deficient mice.45 IL-1α-deficient mice had significantly higher levels of non-high-density lipoprotein cholesterol. The beneficial effect of IL-1α deficiency was due to hematopoietic cells transferred from the bone marrow of IL-1α-deficient mice, resulting in a reduction in aortic lesion size twice that observed in mice transplanted with IL-1β-deficient bone marrow cells. Therefore, IL-1α appears to play a greater role in the pathogenesis of lipid-mediated atherogenesis than IL-1β, and this may be due to an effect of membrane IL-1α.
INTERLEUKIN-1β
Interleukin-1β: The Master Cytokine in the Interleukin-1 Family
More than any other member of the IL-1 family, IL-1β has been the focus of most studies. IL-1β is a highly inflammatory cytokine, particularly in humans, as reviewed in Dinarello.46 As shown in Figure 26.2A, IL-1β and IL-1α bind to the same IL-1RI and trigger a proinflammatory signal. The interest in IL-1β is also due, in part, to it being a secreted cytokine from macrophages and to the importance of the macrophage in antigen presentation before the era of dendritic cells. The inactive IL-1β precursor is converted into an active cytokine by the intracellular cysteine protease caspase-1. In particular, persons with activating mutations in one of the key genes that control the activation of caspase-1 can develop life-threatening systemic inflammation, which is reversed by either blocking the IL-1 receptor or through the use of a neutralizing antibody to IL-1β. Other chronic inflammatory diseases are mediated by IL-1β, as neutralizing antibodies have been used to treat a broad spectrum of diseases.
The IL-1β-mediated illnesses fall into the category of “autoinflammatory” diseases, which are to be distinguished from the classic “autoimmune” diseases. Although inflammation is common to both autoinflammatory and autoimmune diseases, in the case of IL-1-mediated disease, there is no evidence for role of adaptive immunity in its induction.
Interleukin-1β is an Inducible Cytokine
Unlike IL-1α, the IL-1β precursor is not present in health. Also unlike IL-1α, IL-1β is primarily a product of monocytes, macrophages, and dendritic cells, as well as B-lymphocytes and NK cells. In health, circulating human blood monocytes or bone marrow cells do not constitutively express mRNA for IL-1β. Endothelial cells, skin keratinocytes, fibroblasts, and epithelial cells contain constitutive IL-1α and constitutive IL-33 as precursors as well as mRNA, but these cells do not express IL-1β mRNA even upon stimulation with TLR ligands. Melanoma cells do express IL-1β as a precursor, and the more aggressive and metastatic the melanoma, the greater the likelihood of active caspase-1 and IL-1β secretion.47 In the bone marrow neutrophil precursors, IL-1β gene expression is inducible but mature neutrophils in the circulation no longer produce IL-1β. Neutrophil IL-1β plays a pathologic role in the severe inflammation of mice with a mutant form of the phosphatase SHP1.48 Several malignant tumors do express IL-1β as part of their neoplastic nature, particularly acute myelogenous leukemia, melanoma, multiple myeloma, and juvenile myelogenous leukemia, each of which exhibit constitutive expression of IL-1β. Unlike most cytokine promoters, IL-1β regulatory regions are distributed over several thousand base pairs upstream from the transcriptional start site. In addition to a cAMP response element, there are NF-κB-like and activating protein-1 sites. IL-1β gene regulation has been reviewed in detail.49 Although steady-state mRNA levels for IL-1β may be present, there is distinct dissociation between transcription and translation of the IL-1β precursor. Non-TLR ligands such as the complement component C5a, hypoxia, adherence to surfaces, or clotting of blood induce the synthesis of large amounts of IL-1β mRNA in monocytic cells without significant translation into the IL-1β protein. In these cells, the IL-1β mRNA assembles into large polyribosomes, but there is no significant elongation of the peptide.50 This failure to complete the translation into IL-1β protein may be due to the instability element present in the coding region. This instability region is also found in IL-18 and IL-37, and appears to limit the mRNA of these cytokines.51 However, completion of translation of the mRNA into the respective cytokines can be accomplished by adding low concentrations of TLR ligands or IL-1 itself to the “primed” monocytes.52
Processing and Secretion of Interleukin-1β via the Caspase-1
Nearly all microbial products induce IL-1β via TLR activation; in addition, IL-1 (either IL-1α or IL-1β) induces itself both in vivo and in monocytes in vitro.53 Other studies supporting this concept of IL-1-induced IL-1 have been reported.54,55,56,57 Regardless of the stimulus, processing and secretion of IL-1β requires conversion of procaspase-1 to active caspase-1, although in some studies processing of the IL-1β precursor is caspase-1 independent.58 The activation to active caspase-1 is dependent on a complex of intracellular proteins termed the “inflammasome” by the late Juerg Tschopp.59,60 The critical component of the inflammasome is NACHT, LRR, and PYD domains containing protein 3 (NLRP3). NLRP3 is also termed cryopyrin as the gene was initially discovered in patients with “familial cold autoin-flammatory syndrome,” a genetic disease characterized by constitutional symptoms, fevers, and elevated acute-phase proteins following exposure to cold.61
As monocytes exit the bone marrow, they circulate in the bloodstream for approximately 3 days. In the absence of disease, it is likely that these cells do not enter tissues but are destroyed in the spleen or undergo apoptosis. There is no dearth of reports that circulating human blood monocytes release processed IL-1β upon stimulation starting 4 hours after stimulation with TLR agonists and continue to release the cytokine during the following 20 to 40 hours. Following lipopolysaccharide (LPS), IL-1β mRNA levels rise rapidly within 15 minutes but begin to decline after 4 hours due to the short half-life of their mRNA or the action of micro RNA. In contrast, using IL-1 itself as a stimulant, IL-1β mRNA levels are sustained for over 24 hours.52 Raising intracellular cAMP levels with histamine enhances IL-1-induced IL-1 gene expression and protein synthesis. Monocytes of patients with autoinflammatory diseases such as cryopyrinassociated periodic syndrome (CAPS) and hyper IgΔ syndrome (HIDS) release IL-1β even without TLR stimulation during a 24-hour incubation.62,63
When obtained from the venous blood of healthy subjects, human blood monocytes contain active caspase-1. Active caspase-1, as determined by its cleavage into the active dimer, is present even in the absence of stimulation.64 Active caspase-1 present in freshly obtained monocytes is nevertheless dependent on the presence of the key components of the inflammasome, namely ASC and NLRP3.64 However, during subsequent incubation, extracellular levels of adenosine triphosphate (ATP) increase in the supernatant as IL-1β also increases and inhibition of ATP by oxidized ATP reduces the secretion of IL-1β.64 The inhibition of IL-1β secretion by oxidized ATP is consistent with the role of the P2X7 receptor, which binds ATP and opens the potassium channel for release of intracellular potassium. The presence of active caspase-1 in circulating blood monocytes suggests that the rate limiting step in the processing and release of IL-1β is at the level of gene expression.
However, upon differentiation of the same blood monocytes into macrophages in vitro, TLR-induced IL-1β release requires activation of caspase-1 by exogenous ATP.64 The assembly of the inflammasome components with inactive pro-caspase-1 takes place following a fall in intracellular potassium triggered by ATP binding to the P2X7 receptor. ATP activation of the P2X7 receptor opens the potassium channel, and simultaneously, as potassium levels fall, caspase-1 is activated by the inflammasome.65,66,67,68,69 Without exogenous ATP, there is little or no processing of the IL-1β precursor in differentiated monocyte-derived macrophages. Alveolar macrophages obtained from the lungs of healthy human also do not release IL-1β with LPS stimulation unless exogenous ATP is added.64 In addition to ATP activation of P2X7, activation of IL-1β processing can also take place with a cathelicidin-derived peptide termed LL37, which is released from neutrophils.69
The cleavage of the IL-1β precursor by active caspase-1 can take place in the specialized secretory lysosomes or in the cytoplasm. However, more than one pathway seems available for processed IL-1β to exit the cell. These include by exocytosis of the secretory lysosomes,65,66 shedding of plasma membrane microvesicles, and direct release via transporters or multivesicular bodies containing exosomes.70 In general, the release of processed IL-1β takes place before there is significant release of lactate dehydrogenase,71 although in vitro cell death eventually takes place. Pyroptosis is a caspase-1-dependent form of cell death and is induced by certain bacteria using Ipaf, a member of the Nod-like receptor (NLR) family of intracellular receptors.72 An increase in intracellular calcium is also required for the mature IL-1β to exit the cell and is phospholipase C dependent.66
Gain of Function Mutation in Cryopyrin
Diseases associated with single amino acid activating mutations in cryopyrin are termed CAPS. In monocytes from patients with CAPS, activation of caspase-1 occurs without a requirement for a rapid fall in the level of intracellular potassium.57 Therefore, mutated cryopyrin allows for the assembly of the complex of interacting proteins in the presence of normal intracellular levels of potassium. Although often studied using LPS-induced synthesis of the IL-1β precursor,73 it is unlikely that LPS plays a role in autoinflammatory diseases. On the other hand, spontaneous secretion of IL-1β from monocytes of patients is due to endogenous IL-1β stimulation. In patients with CAPS, there is a decrease in steady state levels of pro-caspase-1 mRNA with IL-1Ra treatment,54 suggesting that IL-1β stimulates its own production and processing. Thus, in any disease process that includes an increase in the steady state levels of pro-caspase-1 mRNA, components of the inflammasome or the IL-1β precursor explain the “autoinflammatory” nature of the disease. Type 2 diabetes appears to be an example of an autoinflammatory disease where glucose induces IL-1β production from the insulin-producing beta cell and IL-1β induces the beta cell to produce its own IL-1β.74
Polymorphisms in P2X7 and the Activation of the Inflammasome
Patients with classic autoinflammatory diseases such as Familial Mediterranean Fever (FMF) or CAPS have nearly identical clinical parameters, secrete more IL-1β, and respond dramatically to IL-1 receptor blockade yet have no mutation in NALP3. It is therefore possible that mutations in P2X7 itself or regulation of the other genes controlling potassium channels75 may account for dysfunctional secretion of IL-1β. For example, monocytes from patients with rheumatoid arthritis are more sensitive to release of IL-1β following ATP activation of the P2X7 receptor compared to monocytes from healthy controls.76 However, monocytes from subjects with a P2X7 Glu496Ala loss-offunction polymorphism secrete significantly less IL-1β.77 Monocytes from subjects homozygous for this polymorphism also released significantly less IL-18.78 Another P2X7 receptor polymorphism is associated with increased mortality in patients undergoing allogenic stem cell transplantation.79 Bacteremia was documented in 68% of patients with this polymorphism compared to 18% in wild-type control patients.79
In mice deficient in P2X7 receptors, inflammation, pain, and IL-1β-mediated IL-6 production are markedly reduced.80 In addition to a fall in intracellular potassium, ATP triggers formation of peroxynitrite, which is required for caspase-1 activation because peroxynitrite scavengers prevent IL-1β secretion.81 Pannexin-1, a mammalian protein that functions as a hemichannel for the uptake of dyes, is required for caspase-1 processing and release of IL-1β via the P2X7 receptor.82 Pannexin-1 can also function for LPS-induced IL-1β synthesis in the absence of TLR4.83 P2X7 receptor activity is also regulated by “regeneration and tolerance factor.”84
Non-Caspase-1 Processing of Interleukin-1β
Non-caspase-1 mechanisms also exist to generate active forms of IL-1β. For example, sterile inflammation induces fever, elevated IL-6, and increased production of hepatic acute-phase proteins. These responses are absent in mice deficient in IL-1β but present in mice deficient in caspase-1.85,86 Sterile inflammation is often associated with neutrophilic infiltration and neutrophils produce IL-1β. Because neutrophils are short-lived cells dying within hours upon emigration, release of the IL-1β precursor from intracellular stores is not unexpected. Processing of the IL-1β precursor extracellularly into an active cytokine has been reported for the common neutrophil protease, proteinase-3.86,87 Proteinase-3 also contributes to the processing of IL-18.88 Other proteases such as elastase, matrix metalloprotease 9, and granzyme A process the IL-1β precursor extracellularly. In addition, a mast cell chymase generates active IL-1β.
Mice with a targeted IKKβ deletion in myeloid cells are more susceptible to LPS-induced shock than control mice,55 and markedly elevated levels of IL-1β are found in the circulation associated with a prominent neutrophilia.55 The elevated levels of IL-1β are lethal as blockade with IL-1Ra protects these mice from death. The source of the IL-1β in these mice is the neutrophil. When incubated with proteinase-3, cleavage of the IL-1β precursor is observed yielding molecular weights of 25,000 and 15,000 Daltons.55 Because the cleavage of the IL-1β precursor by proteinase-3, elastase, and cathepsin G are within three amino acids of the caspase-1 cleavage site, the products of the non-caspase-1 cleavage are biologically active.86,87 Therefore, in inflammatory conditions such as urate crystal arthritis, which is characterized by a prominent neutrophilic infiltration, proteinase-3 cleavage of extracellular IL-1β precursor likely takes place.89 Mice deficient in caspase-1 are not protected against urate-induced inflammation. Although IL-1Ra is effective in treating gout, IL-1Ra would be equally effective in any disease with extracellular processing of the precursor.90,91,92 The importance of extracellular processing of the IL-1β precursor by serine proteases may explain, in part, the anti-inflammatory properties of alpha-1-antitrypsin.93
Reactive Oxygen Species and Interleukin-1β Processing
Is there a role for reactive oxygen species (ROS) in the activation of the IL-1β inflammasome? It was reported that uric acid crystals added to human monocytes result in the generation of ROS, which bind to and activate NLRP3 with subsequent secretion of IL-1β.94 However, mice deficient in ROS production exhibit a proinflammatory phenotype.95 Humans with chronic granulomatous disease (CGD) due to mutations in p47-phox cannot generate ROS and are severely affected by inflammatory granuloma. Uric acid crystal activation of primary monocytes from persons with CGD produced fourfold higher levels of IL-1β compared to monocytes from unaffected persons.96 In contrast to previous studies,94 the small molecule ROS inhibitor diphenyleneiodonium, which reduces the production of IL-1β, does so due to inhibition of IL-1β gene expression rather than decreased caspase-1 activation.96 Another study identified phagocyte oxidase-defective monocytes from CGD patients as a source of elevated IL-1β.97 These findings support the concept that ROS likely dampens inflammasome activation and may explain the presence of an inflammatory phenotype characterized by granulomas and inflammatory bowel disease occurring in patients with CGD. In fact, patients with CGD-related inflammatory bowel disease improve upon IL-1 receptor blocking therapy.98
Effects in Mice Deficient in Interleukin-1β
After 10 years of continuous breeding, mice deficient in IL-1β exhibit no spontaneous disease. However, upon challenge, IL-1β-deficient mice exhibit specific differences from their wild-type controls. The most dramatic is the response to local inflammation induced by a subcutaneous injection of an irritant. Within the first 24 hours, IL-1β-deficient mice do not manifest an acute-phase response, do not develop anorexia, have no circulating IL-6, and have no fever.85,99 These findings are consistent with those reported in the same model using anti-IL-1R type I antibodies in wild-type mice.85,99 IL-1β-deficient mice also have reduced inflammation due to zymosan-induced peritonitis.85,100 In contrast, IL-1β-deficient mice have elevated febrile responses to LPS, IL-1β, or IL-1α compared to wild-type mice.101 Nevertheless, IL-1β-deficient mice injected with LPS have little or no expression of leptin mRNA or protein.102
Mice deficient in IL-1β were compared to mice deficient in IL-1α after exposure to chemical carcinogens.103 In IL-1β-deficient mice, tumors developed slower or did not develop in some mice. A deficiency in IL-1α, on the other hand, did not impair tumor development compared to wild-type mice injected with the same carcinogen. In IL-1Ra-deficient mice, tumor development was the most rapid. A leukocyte infiltrate was found at the site of carcinogen injection. The neutrophilic infiltrate was almost absent in IL-1β-deficient mice, whereas in IL-1Ra-deficient mice, a dense neutrophilic infiltrate was observed. In wild-type mice, the leukocytic infiltrate was sparse and the infiltrate that was observed in IL-1α-deficient mice was similar to that of control mice. These findings may reflect the fact that IL-1β is secreted into the microenvironment resulting in the emigration of monocytes and neutrophils, whereas IL-1α remaining cell-associated is less likely to affect the microenvironment.
Interleukin-1α and Autophagy
Autophagy is an ancient process of recycling cellular components, such as cytosolic organelles and protein aggregates, through degradation mediated by lysosomes. Autophagy is activated in conditions of cell stress, hypoxia, starvation, or growth factor deprivation; it promotes cell survival by generating free metabolites and energy through degradation of the endogenous cellular components.104 However, in addition to its role in the pathophysiology of cancer, neurodegenerative diseases, or aging, autophagy is also a modulator of inflammation.105 A role for autophagy in production of proinflammatory cytokines, particularly of IL-1β, has emerged with deletion of ATG16-L1. For example, macrophages from ATG16L1 -deficient mice produce higher levels of IL-1β and IL-18 after stimulation with TLR4 ligands.106 The data suggest that higher activation of caspase-1 in the ATG16L1-deficient mice accounts for the higher production level.106 This observation was related to the specific degradation of the IL-1β precursor in autophagosomes in mouse macrophages.107 Additional studies in the ATG16L1-deficient mice point toward a regulatory effect of autophagy on caspase-1 activation through modulation of the NLRP3 inflammasome.94,108,109
This role of autophagy in the secretion of IL-1β was also observed in human primary monocytes, in which specific inhibition of autophagy leads to increased production of IL-1β.110 However, in the same cells, tumor necrosis factor (TNF)α production was decreased by autophagy inhibition. These data suggest divergent effects of autophagy on the production of these two important proinflammatory cytokines. In mice, the increase in IL-1β production is ascribed to increased activation of the inflammasome, but in human cells, it is IL-1β mRNA transcription that is elevated when autophagy was inhibited, whereas no effects were observed on caspase-1 activation.106,107,110 Despite these differences between mouse and human cells, the inhibition of autophagy increases the production of IL-1β but not TNFα.
The modulation of inflammation by autophagy in humans has been studied in Crohn disease. Genome-wide association studies in large cohorts of patients with Crohn disease have revealed that genetic variants in two autophagy genes, ATG16L1 and IRGM, result in increased susceptibility to the disease. A nonsynonymous polymorphism in ATG16L1 on chromosome 2q37.1 and two polymorphisms in IRGM on chromosome 5q33.1 were significantly associated with Crohn disease risk.111,112 Another study revealed a significant association of Crohn disease susceptibility with an intronic polymorphism in the autophagy gene ULK1.113 Moreover, autophagy defects have been reported in individuals bearing nucleotide oligomerization domain (NOD)2 mutations and are consistent with the concept that impaired bacterial clearance and increased bacterial persistence are part of the pathogenesis of Crohn disease.114
The mechanism through which polymorphisms in autophagy genes influence susceptibility to Crohn disease appears to involve IL-1β production. The ATG16L1 300Ala risk allele was associated with elevated production of IL-1β and IL-6; however, this finding was only observed in cells stimulated with the NOD2 ligand muramyl dipeptide. In contrast, the expected levels of IL-1β and IL-6 were produced upon stimulation with TLR2 and TLR4 ligands.115 The increased production of IL-1β was associated with an increase in the steady state levels of IL-1β mRNA rather than increased activation of the inflammasome.115 Studying the same polymorphism (ATG16L1 Thr300Ala) in human dendritic cells, Cooney et al. reported defective NOD2-induced, but not TLR-induced, autophagy and antigen presentation.116 Furthermore, effects of this polymorphism on antibacterial autophagy in epithelial cells have been observed.117 The specific effect of the ATG16L1 polymorphism on the NOD2 pathway, and not on TLR-induced stimulation, is likely related to the fact NOD2 and ATG16L1 form a protein complex that is essential for NOD2-induced autophagosome formation.118 Because the ATG16L1 Thr300Ala polymorphism affects protein stability,119 defective induction of autophagy and therefore enhanced IL-1β mRNA transcription upon triggering of NOD2 may be due to the presence of defective complex.
INTERLEUKIN-33
Interleukin-33 as a Member of the Interleukin-1 Subfamily
Formerly termed IL-1F11, IL-33 belongs to the IL-1 subfamily and has been studied for its role in the Th2 paradigm of immune responses. IL-1β is also linked to the Th2 response. The existence of IL-33 was predicted in 1994 following the discovery of a novel member of the IL-1 receptor family termed ST2.120 ST2 is the ligand binding chain for IL-33 (Table 26.2) and is structurally similar to the ligand binding chain of IL-1α and IL-1β. In addition, the coreceptor for IL-33 is the IL-1RAcP, which is also the coreceptor for IL-1α and IL-1β. It was not until 2005 that IL-33 was reported as the ligand for ST2.11 ST2 is regulated by the estrogen inducible transcription factor Fos,120 and this property of estrogens may be related to the large number of studies on the effect of estrogens to regulate IL-1 and inflammation.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


