The Innate Immune System
Luke A. J. O’Neill
INTRODUCTION
Every organism has to contend with infection by microbes. Infectious agents such as bacteria, viruses, and fungi threaten health and viability by competing for nutrients or more dangerously by killing the infected host as a consequence of infection or in order to spread to another host. This threat occurs largely because microorganisms divide at a faster rate than the host and because of the production of toxins that the microorganism uses for its own viability. The job of the immune system is firstly to sense the danger posed and secondly to mount an appropriate response. This response can be to tolerate the microorganism or to resist it such that at best sterilizing immunity occurs.
The immune system is traditionally split into two categories: innate and acquired (or adaptive) immunity. The term “innate” refers to the feature that the sensors involved are encoded by genes that do not undergo any rearrangement to generate variants. Adaptive immunity, on the other hand, refers to host defense proteins encoded by genes that undergo rearrangement to generate great diversity. This rearrangement process is not driven by the infectious agent, but happens by genetic programming. Antigens then lead to an expansion of antigen-specific B cells or T cells that help clear the infection and importantly persist in the form of memory cells. This means that when the host is re-infected, there are substantially greater numbers of the specific B- and T-cell populations that lead to clearance of the infectious agent. There are therefore insufficient numbers of microbes to trigger substantial systemic symptoms.
It is estimated that more than 99% of life on earth only has innate immunity.1 What advantage would the evolution of adaptive immunity bestow on an organism? The answer lies in part in the nature of innate immunity. As will be discussed, the effector mechanisms here involve the inflammatory process, which when out of control can harm the host. In mammals, a “sickness behavior” also occurs, which includes processes such as fever, aches and pains, and drowsiness, which are thought to allow the infected host to rest and recuperate.2 They also present a disadvantage, however, because they leave the host vulnerable to predators and decrease reproductive potential. Adaptive immunity occurs later in the cycle of infection, typically becoming evident several days after infection, and probably only after innate immune defenses have been breached. It is needed for sterilizing immunity. The fact that memory is a key feature of adaptive immunity, however (which is evident because of the expansion of specific B and T cells), means that upon re-infection, the innate response is limited because the pathogen is more rapidly cleared and there are therefore fewer microbial products present to activate innate immunity. In addition, there is evidence that adaptive immune cells actually inhibit the innate immune response, which in the first infection acts in a negative feedback manner.3 In subsequent infections, the memory cells probably inhibit innate immunity from the start of the infection. In essence, therefore, it appears that adaptive immunity evolved to promote sterilizing immunity and also to prevent innate immunity happening upon reexposure to pathogen, given its potential disadvantages in terms of systemic inflammation. Figure 15.1 illustrates this principal.
Previously thought to be crude and nonspecific, innate immunity is now known to involve a range of receptor families that recognize diverse microbial products. Importantly, these receptors program gene expression changes in target cells. The products of these genes drive inflammation and are also required for adaptive immunity. These proteins include cytokines that promote expansion of B and T cells. Other innate immune proteins have direct antimicrobial effects or act as opsonins, coating bacteria and promoting phagocytosis. Innate immunity has therefore gone from being viewed as a process lacking sophistication, to one determining for both inflammation and adaptive immunity.4
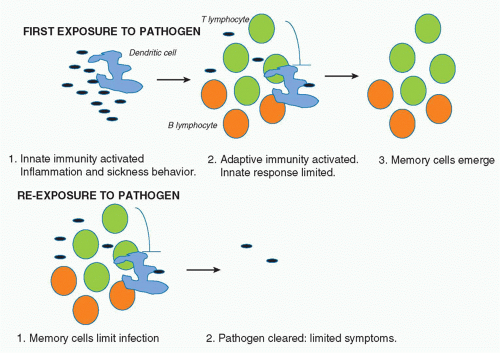 FIG. 15.1. The Relationship between Innate and Adaptive Immunity. A: Upon exposure to a pathogen, the innate immune response is triggered mainly by phagocytes and dendritic cells. Inflammation ensues, which leads to sickness behavior and host defense. The adaptive immune system is then triggered leading to effector T- and B-lymphocytes, some of which become memory cells. Adaptive immunity also has a negative feedback effect on innate immunity, preventing damage. B: When an organism is exposed to the same pathogen again, the memory cells can handle the infection, limiting innate immunity such that there are only minor symptoms. The evolution of adaptive immunity therefore provides an advantage to the host: adaptive immunity evolved to clear pathogens and limit innate immunity. |
THE FRONTLINE OF INNATE IMMUNITY
The Skin
Innate immunity at its simplest involves barriers that prevent entry of microorganisms in the first place. In mammals, the skin is an obvious barrier that when broken in injury allows pathogens to enter. Many pathogens use insects that can deliver them across the skin into the bloodstream. A good example here is Plasmodium falciparum, which causes malaria. It is transmitted by mosquitoes feeding on blood. If the skin is broken, pathogens can enter. The importance of the integrity of the skin barrier can be seen in atopic dermatitis, an inflammatory skin disease. Mutations in the gene encoding fillaggrin, which is important for skin integrity, associate with atopic dermatitis.5 The altered fillaggrin is less able to maintain the skin barrier, and the dermatitis is likely to be caused by microorganisms penetrating the skin or environmental irritants that provoke allergy.
The Epithelial Barrier
Equally important is the epithelial barrier that lines surfaces inside the body in the respiratory, gastrointestinal, and urogenital tracts. Microbes gain access to these surfaces via processes such as inhalation and digestion. The epithelial cells that make up the epithelium form tight junctions that keep the microorganisms out.6 Specialized structures called cilia also expel microbes, and damage to cilia, for example caused by cigarette smoke, promotes respiratory tract infections.7
DIRECT-ACTING ANTIMICROBIAL FACTORS
The epithelia also produce a range of chemicals that limit microbial growth. Mucus is a key substance that traps microbes and is expelled. The importance of mucus can be seen in cystic fibrosis, where unusually thick mucus is made in lungs that limits expulsion and gives rise to growth of bacteria in the airways.8 This chronic infection in turn leads to the lung pathology in this disease. In the stomach, acid production by the epithelium leads to low pH that limits growth of microbes. Enzymes that digest microbial proteins are also produced, including pepsin in the gut. A phospholipid-based substance termed pulmonary surfactant is made in the lungs that acts to trap microbes and can also directly lyse bacteria. Similarly, in skin the production of fatty acids in sebum limits bacterial growth. Lysozyme is a prominent enzyme in tears and saliva that breaks down peptidoglycan in the cell walls of gram-positive bacteria. Lysozyme is in fact a prototype of an innate immune protein because it does not affect the host cells, as they lack peptidoglycan. It is made by epithelial cells, but also phagocytes and Paneth cells in the crypts in the small intestine.
Antimicrobial Peptides
Another important example of innate immune proteins produced at barriers is antimicrobial peptides.9 Epithelial cells are a major source of these peptides, but phagocytes can also make them. They are an ancient form of defense because they are found in most species, including insects and plants. There are three classes: defensins, catelicidins, and histatins. Defensins are 30 to 40 amino acid peptides that usually have three disulphide bonds. Their key property is amphipathicity (they contain both polar and nonpolar properties), and it is this property that is thought to underly their specificity for the membranes of bacteria, fungi, and certain viruses. Their hydrophobic part inserts into these membranes, which are more positively charged than host cell membranes. A pore is formed, rendering the membrane leaky and leading to lysis.
There are three subfamilies of defensins-: α, β, and θ, which have different specificities in terms of target microorganisms.10 They are produced as propeptides that require processing by proteases. Neutrophils produce α-defensins and store them in so-called primary granules from which they are secreted upon neutrophil activation. Paneth cells in the gut produce a class of α-defensins called cryptidins, which are processed by trypsin prior to secretion into the gut., β-defensins are made in the urogenital and respiratory tracts, skin, and tongue. Keratinocytes in skin are also a key producer of β-defensins.
There are three subfamilies of defensins-: α, β, and θ, which have different specificities in terms of target microorganisms.10 They are produced as propeptides that require processing by proteases. Neutrophils produce α-defensins and store them in so-called primary granules from which they are secreted upon neutrophil activation. Paneth cells in the gut produce a class of α-defensins called cryptidins, which are processed by trypsin prior to secretion into the gut., β-defensins are made in the urogenital and respiratory tracts, skin, and tongue. Keratinocytes in skin are also a key producer of β-defensins.
Catelicidins lack the disulphide bonds in the α-defensins and are produced by epithelial cells, phagocytes, and keratinocytes. They are also produced as propeptides and in neutrophils are processed by elastases in secondary granules and then secreted in response to microbial stimulation. Histatins are produced in the parotid, sublingual, and submandibular glands in the oral cavity. They are histidine-rich and target fungal membranes.
Phospholipase A2
Phospholipase A2 (PLA2) is another antimicrobial protein found in primary granules of neutrophils and similar to defensins, is secreted during degranulation in response to bacterial activation. It is also secreted by epithelial cells and Paneth cells. PLA2 can directly kill gram-positive bacteria by hydrolysing phospholipids in the microbial membrane.11 The means by which it distinguishes host membranes from bacterial membranes is because its effect on phospholipids is strongly potentiated by another protein termed bactericidal permeability increasing (BPI), which binds to lipopolysaccharide (LPS) in gram-negative bacterial membranes.12 As will be discussed subsequently, LPS is one of most potent innate immune activators known; it is sensed by a protein termed toll-like receptor-4 (TLR4). LPS does not occur in host membranes, and so BPI only facilitates the effect of PLA2 on bacterial membranes. BPI disrupts the integrity of the bacterial membrane and presumably allows PLA2 to gain access to phospholipids, where it cleaves fatty acids from the sn-2 position, leading to lysis. Similar to PLA2, BPI is found in primary granules in neutrophils.
Lactoferrin
Lactoferrin is an 80 kDa protein that belongs to the transferrin family and therefore acts to sequester iron. This effect is thought to limit the availability of iron to bacteria, altering their metabolism and thereby compromising their ability to replicate.13 Lactoferrin is found in the secondary granules of neutrophils and occurs at a high level in breast milk, its cellular origin there being epithelial cells. Lactoferrin can also be processed to generate an antimicrobial peptide called lactoferricin that, similar to other such peptides, can lyse bacterial membranes.
The Microbiota
A final front-line mechanism to keep microbes at bay is commensal bacteria in the healthy intestine. Commensal bacteria live in our intestines and serve several functions, including providing vitamin B12 and other nutrients. However, they also maintain epithelial barrier integrity, most likely because they are sensed by innate immune receptors that will be described in subsequent sections, and these receptors seal the epithelial barrier against infection.14 However, importantly they also compete with any pathogenic bacteria in the intestines limiting their growth. Similarly, on skin and in the urogenital tract, there are competing microbes.
THE INITIAL RESPONSE ONCE BARRIERS ARE BREACHED
What happens when skin or epithelial barriers are breached? Pathogens have mechanisms to get across the barriers, or if the barrier is damaged by trauma, pathogens will traverse the broken barrier. Two important cascades are triggered in blood that will generate inflammatory factors or act to wall off pathogens and prevent them from spreading. The first cascade to be characterized in detail is the complement cascade, described in detail elsewhere in this volume. Complement generates the membrane attack complex that will lyse bacteria. Complement factors such as C1q also bind to pathogen surfaces and act as opsonins, promoting uptake by neutrophils and phagocytes. The complement cascade also generates chemotactic factors such as C5a, which attracts neutrophils to the site of infection. Coagulation is the other cascade that is triggered, largely by bacteria. This will form clots that prevent bacteria from spreading.
The Neutrophil
A key cell type, however, that is engaged early in infection is the neutrophil, again described in detail elsewhere. Chemotactic peptides present in bacteria, termed fMLP, act as chemoattractants for neutrophils. Both C5a and fMLP also trigger an important process in the neutrophil termed the respiratory burst.15 This involves an enzyme complex termed the nicotinamide adenine dinucleotide phosphate-oxidase. This is also present in macrophages and is activated either by the G-protein coupled receptors that bind fMLP or C5a, or by the process of phagocytosis. It is a multicomponent enzyme complex that contains three cytosolic subunits: p40phox, p47phox, and p67phox. It also contains a membrane-associated flavocytochrome complex comprising p22phox and p91phox. The subunits form the enzyme complex following activation of the low molecular weight G protein Rac. The nicotinamide adenine dinucleotide phosphate-oxidase generates reactive oxygen species by transferring an electron from its flavin adenine dinucleotide cofactor to molecular oxygen forming the superoxide anion and other oxygen radicals. This all occurs in the so-called phagolysosome, which comprises a fusion between the phagosome that contains the ingested bacteria with the lysosome. The oxidizing environment will be antibacterial, but in addition, will lead to acidification of the phagolysosome due to potassium and hydrogen ions being drawn in to neutralize the charged superoxide ion. The acidification, however, has been shown to lead to dissociation of proteolytic
enzymes such as elastase from the proteoglycan matrix in the phagolysosome.16 These proteases will also have a direct effect on bacteria.
enzymes such as elastase from the proteoglycan matrix in the phagolysosome.16 These proteases will also have a direct effect on bacteria.
In addition, superoxide is a substrate for myeloperoxidase, which is stored in primary granules and generates hypochlorous acid and chloramines, as well as other reactive oxygen species that are bactericidal and fungicidal. A final oxygen radical generated in neutrophils is nitric oxide. It is produced by the enzyme nitric oxide synthase, which is induced by cytokines such as interferon-γ. Nitric oxide is also microbicidal.
THE CONCEPT OF PATTERN RECOGNITION: HISTORICAL PERSPECTIVE
Much of the forgoing components of innate immunity had been worked out at least in broad terms by the mid-1990s. However, the issue of the exact recognition mechanism during innate immunity by the host was poorly understood. In 1989, Janeway made the point that the continued focus on adaptive immunity, which was evident at the time, would lead to an asymptote being approached in our understanding of immunity.17 He speculated that receptors must exist for microbial structures that were known to be proinflammatory, such as LPS from gram-negative bacteria. Receptors such as mannan-binding lectin (MBL), which binds mannose in microbial membranes (and which does not exist in host membranes) and CD14 (which binds LPS), had already been described at that stage, but importantly these proteins did not signal because they possessed no intracellular domains. MBL was shown to be an opsonin, and CD14 had no clear function. Janeway therefore proposed that pattern recognition receptors (PRRs) must exist that recognize microbial “patterns” such as LPS, termed pathogen-associated molecular patterns (PAMPs), and trigger activation of cells such as dendritic cells, which are prodigious producers of cytokines and also present antigen to T cells. At around the same time, Matzinger was proposing that the job of the immune system is to sense “danger.”18 The danger can be in the form of a microbe or in the form of tissue injury. Again, the idea that specific receptors would exist to recognize products of damaged tissue was proposed. In essence, what was being hypothesized was the existence of receptors that would drive the production of cytokines, as several cytokines had been found that could provoke inflammation and also adaptive immunity. LPS was a powerful inducer of such cytokines and yet no receptor had been convincingly described for LPS that would activate transcription factors such as NF-kB, which was known to be required for increased transcription of genes such as that encoding the potent proinflammatory cytokine tumor necrosis factor (TNF).
How did Janeway’s insight that PRRs and PAMPs would form the core of innate immunity lead to the discovery of the TLRs, which became the prototypical PRRs? Five important findings were made.
The receptor for the proinflammatory cytokine interleukin (IL)-1, which is a key mediator of many aspects of inflammation and which, similar to LPS, could activate NFκB, was shown to have a signalling domain similar to a Drosphila melanogaster protein Toll.19 The Toll protein had been discovered as being important in the generation of dorsal-ventral polarity in the developing Drosophila embryo. At first glance, this seemed odd. What was a receptor important for inflammation doing with a signaling domain highly similar to a protein in fruit fly development? Toll drives a transcription factor termed dorsal, however, which is highly similar to NFκB so it seemed as if a similar “machine” was being used in two different contexts: one for inflammation in the case of IL-1 and the other for development in the fruit fly.
In 1994, a protein termed N protein was described in tobacco by Whitham and colleagues, which provided resistance to tobacco mosaic virus.20 This protein also had the domain found in the type I IL-1 receptor and Toll, which was then named the Toll-IL-1 receptor -resistance (TIR) domain. Clearly, the TIR domain was therefore important for innate immunity in very diverse species.
This was confirmed when in 1996, Lemaitre and colleagues reported that Toll also has a role in innate immunity in Drosophila, being activated in response to fungi and driving the antifungal peptide drosomycin.21
Mammalian proteins even more similar to Toll than the IL-1 receptor, and termed TLRs, were then reported22 and one, termed Toll (subsequently renamed TLR4) when overexpressed in dendritic cells, was shown to drive costimulation of T cells, as reported by Medzhitov and colleagues.23
In 1999, Poltorak and colleagues reported that the lack of responsiveness of a strain of mouse termed C3H/HeJ to LPS was due to a mutation in the gene encoding TLR4, such that a proline in the signaling domain was converted to a histidine.24 This rendered TLR4 unable to signal, identifying TLR4 as the longsought signaling receptor for LPS. It was therefore clear that TLRs were ideal examples of the PRRs proposed by Janeway. Subsequently,10 TLRs were described in human, although other species have very many more, notably the sea urchin, which has over 150.25 TLRs were shown to recognize a diverse range of PAMPs and also in some cases products of damaged tissue, termed danger-associated molecular patterns (DAMPs). Complex signaling pathways were elucidated and links to disease established. The discovery of TLRs also galvanized researchers to seek other PRR types, and there are now several families, notably the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), which sense viral ribonucleic acid (RNA), the nucleotide oligomerization domain (NOD)-like receptors (NLRs), which sense bacteria and DAMPs, and the C-type lectin receptors, which sense fungi.26 Figure 15.2 illustrates the main families of PRRs. A level of complexity in innate immunity was therefore revealed that has provided great insight into the overall workings of the immune systems of many organisms.
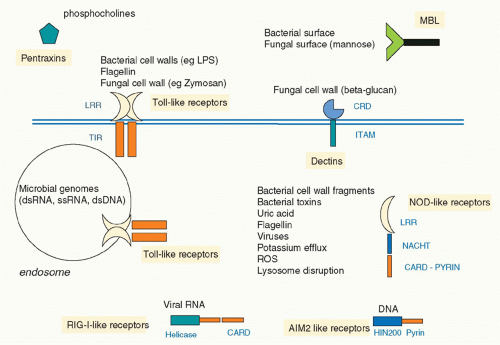 FIG. 15.2. Innate Immune Sensors. Several classes of innate immune sensors have been found. Soluble factors such as pentraxins and mannoan-binding lectins bind pathogen structures and promote phagocytosis. Toll-like receptors (TLRs) sense bacteria, fungi, and protozoans from the plasma membrane, whereas endosomal TLRs sense microbially derived nucleic acids. C-type lectin receptors sense fungal cell wall components and mycobacteria. In the cytosol nucleotide oligomerization domain-like receptors sense microbial products and also, in the case, of Nlrp3 particles such as uric acid and IAPP. Retinoic acid-inducible gene I-like receptor sense viral ribonucleic acid, whereas AIM-2-like receptors sense microbial deoxyribonucleic acid. All lead to signaling pathways culminating in the induction of immune and inflammatory genes. |
Toll-Like Receptors
TLRs are type I transmembrane receptors that have leucinerich repeats (LRRs) and a TIR domain. LRRs are found in many proteins, and that they occur in TLRs is of interest given their capacity to interact with different kinds of ligands. Also of note is that in agnathans, the LRR is used to build a repertoire of variable lymphocyte receptors, which are analogous to the antibody repertoire in human.27 The LRR domain is therefore of evolutionary interest in that it is used for TLRs but also generation of diversity in the lamprey and the hagfish. The immunoglobulin domain serves this purpose in vertebrates and in fact is fused to the TIR domain in the type I IL-1 receptor. Figure 15.3 illustrates the relationship between these domains.
The 10 TLRs in human have different ligand specificities, and several ligand/TLR structures have been solved.28 Broadly speaking, TLRs can be split into two families. TLRs 1, 2, 4, 5, and 6 are expressed in the plasma membrane, and their job is to sense bacterial, fungal, and protozoal products. TLRs 3, 7, and 9 are all expressed in endosomal membranes with their LRRs in the lumen and their TIR domains pointing into the cytosol. Their role is to sense viral nucleic acids. There are also differences in relation to signaling,29 with all of the TLRs with the exception of TLR3 signalling via MyD88, to NF-κB and mitogen-activated protein (MAP) kinases such as p38. TLR2 and especially TLR4 require a bridging adapter termed Mal to recruit MyD88. TLR3 signals via Trif to NF-κB and another transcription factor IRF3. TLR4 can also signal via Trif and does so via the second bridging adapter Tram.
Toll-Like Receptors 2, 1, and 6
TLR2 recognizes multiple ligands. From bacteria, these include lipopeptides, lipoproteins, lipoteichoic acid, and mycobacterial lipoarabinomannan. From yeast, TLR2 recognizes the cell wall structure zymosan. TLR2 also recognizes glycosylphosphoinositol from the parasite Tryanosoma cruzi. Alone amongst the TLRs, TLR2 can heterodimerize with other TLRs, and this provides a degree of specificity in ligand recognition.30 The TLR1/TLR2 dimer binds triacylated lipopeptides whereas TLR2/TLR6 recognizes diacylated lipopepides. In the case of TLR2/6, one acyl chain fits into a hydrophobic pocket in one TLR, whereas the second acyl chain fits into the other TLR.31 This allows the TLRs to dimerize, an important event for signaling. There is also a coreceptor for TLR2/6 in the form of CD36, which appears to deliver the
lipopeptide to the TLR dimer.32 TLR2 is expressed on macrophage, dendritic cells, B cells, and also on regulatory T cells.
lipopeptide to the TLR dimer.32 TLR2 is expressed on macrophage, dendritic cells, B cells, and also on regulatory T cells.
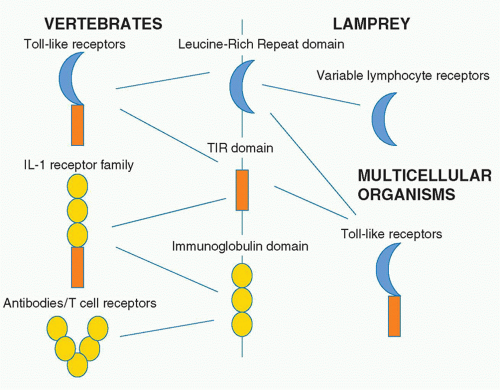 FIG. 15.3. Protein Domains in Innate and Adaptive Immunity. Three protein domains occur in several immune system proteins. The leucine-rich repeat occurs in toll-like receptors in vertebrates and all multicellular organisms combing with the toll-IL-1 receptor-resistance (TIR) domain. The leucine-rich repeat also forms the basis of the repertoire of variable lymphocyte receptors in the hagfish. The immunoglobulin domain only occurs in vertebrates and is found in antibodies and T-cell receptors. However, it also combines with the TIR domain to form interleukin-1 receptor family members. |
Toll-Like Receptor 4
TLR4 is the receptor for LPS and is the most studied TLR. It acts as a homodimer but on its own is unable to recognize LPS. Similar to TLR2 and CD36, a coreceptor is needed, termed MD2.33 LPS is a hexacylated lipid, and five of the acyl chains are buried in MD2, whereas the sixth is on the surface and interacts with TLR4.34 However, two other proteins are needed to deliver LPS to MD2. LBP occurs in serum and binds LPS monomers, transferring them to CD14.35 CD14 can occur as a soluble protein or as a glycerophosphatidylinositol anchored protein.36 The job of CD14 appears to be to concentrate LPS and deliver it to MD2. In B cells, there are two other accessory molecules: RP105 and MD1.37 These appear to serve an analogous function to CD14 and MD2. Recognition of LPS is therefore highly complex. This is presumably because once TLR4 is activated, there can be lethal consequences in the form of septic shock; therefore, several proteins are needed for LPS recognition. Similar to TLR2, a dimerization event occurs upon ligand binding that allows the intracellular TIR domains to come into juxtaposition and trigger signaling pathways. TLR4 has been shown to recognize other ligands, notably F protein from respiratory syncytial virus,38 although the basis for this recognition is not known. Another example is the metal nickel. In contact dermatitis, there is an inflammatory reaction to nickel, and it has been shown that nickel coordinates two key histidines on different TLR4 proteins, causing a cross-linking event that activates TLR4 and drives the inflammatory process in skin.39 TLR4 is expressed on multiple cell types, notably on macrophages and dendritic cells but also on neutrophils and in the endothelium.
Toll-Like Receptor 3
TLR3 is the receptor for double-stranded RNA, found in viruses or more commonly in the form of the viral RNA analogue polyIC. PolyIC had been known for some time to be a potent inducer of the antiviral type I interferons. Similar to LPS, therefore, the discovery of TLR3 as the receptor for polyIC provided a molecular explanation for how polyIC induces this response. TLR3 is expressed on macrophages and dendritic cells and importantly on CD8+ dendritic cells, which are potent type I interferon producers.
Toll-Like Receptor 5
TLR5 is the receptor for bacterial flagella.40 Flagellin is a key protein in flagella, which occur in multiple bacteria species. Flagellin is in many ways a prototypical PAMP because it does not occur in humans and can bear few mutations in bacteria. TLR5 is therefore required for host defence against flagellated bacteria such as Legionella and Salmonella.
Toll-Like Receptor 7
TLR7 recognizes single-stranded RNA from viruses. It is highly expressed in plasmacytoid dendritic cells, where when activated it induces type I interferons. TLR7 was first described as the receptor for a small molecule termed imiquimod, which is used to treat genital warts. This effect is mediated by type I interferons. TLR7’s expression on plasmocytoid dendritic cells reveals a key function in antiviral immunity.
Toll-Like Receptor 9
TLR9 is the deoxyribonucleic acid (DNA) sensing TLR that, similar to TLR3 and TLR7, is found in endosomes. The type of DNA recognized is of interest as it binds to hypomethylated CpG-rich DNA, commonly found in bacteria and viruses. TLR9 is also expressed in plasmacytoid dendritic cells as well as in macrophages and in the epithelium. Once activated, again by dimerization, TLR9 induces a strong proinflammatory signal, increasing TNF production as well as type I interferons. A protein termed Unc93b is required to traffic TLR9 from the Golgi to intracellular vesicles. The CpG-rich DNA
from pathogens is taken up by cells in an endosome, which fuses with the TLR9-expressing endosome. In this way, the receptor can interact with its ligand. TLR9 has no known coreceptors, but it has been shown that it requires cleavage by cathepsins for full activation.41
from pathogens is taken up by cells in an endosome, which fuses with the TLR9-expressing endosome. In this way, the receptor can interact with its ligand. TLR9 has no known coreceptors, but it has been shown that it requires cleavage by cathepsins for full activation.41
The Recognition of Danger-Associated Molecular Patterns by Toll-Like Receptors
TLRs have also been implicated in the recognition of products of damaged tissues.42 This is an important area as the inflammatory process can occur under conditions where there is apparently no infection. DAMPs include a range of factors released by damaged cells and include high-mobility group protein 1, heat shock proteins such as hsp90, products of damaged connective tissue such as hyaluronic acid fragments, versican and tenascin-C, the acute-phase reactant serum amyloid A, pyrrole-based lipids such as CEP, and coagulation products such as fibrinogen. This area has been somewhat controversial, as in vitro there is a risk of LPS contamination. However, the consensus has emerged that at least some of these are indeed sensed by TLRs although what the mechanism might be is not fully understood. Many of these are sensed by TLR4, notably high-mobility group protein-1, hsps, hyaluronan fragments, and tenascin-C. TLR2 has been shown to sense versican and CEP. Importantly, these factors are only produced by damaged cells, as would occur during tissue injury. There may be coreceptors involved here, which explains the recognition process, because it is difficult to imagine how a single TLR can recognize multiple ligands. A clearer example is the sensing of host nucleic acids in the case of cell damage. Necrotic cells have been shown to release double-stranded RNA, which is sensed by TLR3. TLR7 can sense host RNA, and TLR9 can sense host DNA in the context of immune complexes. This has particular relevance to autoimmune diseases such as systemic lupus erythematosus (SLE).43
Toll-Like Receptor Signaling Pathways
The key output from TLR signaling is cytokines, and TLRs filled the missing gap in our understanding of what actually induces cytokines. Because they bind directly to microbial ligands, TLRs bring us close to the initiation of host defense.
The type of cytokine being induced is dependent on the TLR. TLR4 is the most prodigious activator of macrophages known, with many genes induced by TLR4. Other TLRs are less prolific but all share the feature of driving inflammation. Cytokines such as IL-6, TNF, and the chemokine IL-8 are typical responses for plasma membrane receptors. For the endosomal TLRs, type I interferons are the typical readout. What gives rise to signaling specificity? As stated previously, TLRs use different adapter proteins. All have a TIR domain, and upon TLR dimerization, adapter TIR domains are likely to be recruited to the interface formed between two TLRs. This then launches the signaling pathways.
Broadly speaking, TLR signaling pathways can be divided into two: MyD88-dependent and MyD88-independent. All TLRs, with the exception of TLR3, signal via MyD88. IL-1 receptor family members, including the receptor for IL-18, also signal via MyD88.44 The MyD88 dependent pathway involves the IL-1 receptor-associated kinases (IRAKs). There are four of these: IRAK-1, IRAK-2, IRAK-4, and IRAK-M. They interact with MyD88 via their death domains, because along with the TIR domain, MyD88 has a death domain. The structure of this multicomponent signaling machine, termed the MyDDosome, has been solved.45,46 Strikingly, it has a stoichiometry of 14. SIx MyD88 subunits assemble and bring four IRAK-4s to the complex. These sit on top of the assembled MyD88 subunits in a helical structure. Four IRAK-2s or IRAK-4s are then recruited. All of the death domain interfaces in this complex have been solved, and it is predicted to have a high degree of cooperativity. The so-called MyDDosome launches signaling pathways activated by TLRs.
Once assembled, IRAK-4 phosphorylates IRAK-1, and this event appears to be key for signaling. The IRAKs then dissociate from the complex, and IRAK-1 and/or IRAK-2 then interact with the ubiquitin ligase Traf6, a member of the Traf family. Once activated, Traf-6 signals in association with two noncanonical E3 ubiquitin ligases, Ubc13 and Uev1A.47 This results in polyubiquitination of Traf6. This is a type of ubiquitination termed K63-linked ubiquitination. This covalently modifies Traf-6 causing oligomerization and downstream activation. This activation involves the IkappaB kinase (IKK) complex for the NF-κB pathway. The main form of NF-κB in cells is a p50/p65 dimer. It is, however, kept in check by an inhibitory protein I-κB. Traf6 activates the kinase TAK-1, which in turn activates the IKK complex leading to I-κB degradation (via K48-linked polyubiquitination). This releases the p50/p65 dimer, exposing a nuclear localization sequence and allowing NF-κB to bind its consensus site in target genes. In addition, TAK-1 can activate MAP kinase cascades, notably via MKK3/6, which leads to p38 MAP kinase activation, and MKK7, which leads to activation of Jun N-terminal kinase. Both of these MAP kinases can activate transcription factors, such as ATF3 in the case of p38 and Jun in the case of Jun N-terminal kinase. This in turn will modulate inflammatory gene expression.48
MyD88 is also involved in activation of IRF7 by TLR7 and TLR9.49 This is an important transcription factor for the induction of interferon-alpha, a key antiviral cytokine but also a cytokine implicated in the pathogenesis of SLE.
The importance of the MyD88 pathway can be seen in MyD88-deficient mice, which are unable to mount an effective host defense response to multiple pathogens, including a wide range of bacteria, as well as fungi, protozoans, and certain viruses.50 In addition, MyD88-deficient mice are resistant to inflammation in a number of disease models including models of SLE and arthritis. However, in the case of humans, the situation is not as clear-cut. This is because of reports of MyD88 deficiency in human.51 People with this deficiency show higher mortality in childhood but susceptibility to a restricted set of pathogens, the main defect in host defense being against pyogenic infections such as Streptococci. This is also the case in subjects with IRAK-4 deficiencies.52 Once people with these deficiencies reach adulthood, however, no immunodeficiencies are evident. It is likely, therefore, that the MyD88 pathway is somewhat redundant in children and is not required in adults probably
because of the emergence of adaptive immunity. This supports the concept raised at the start of this chapter, that once memory is established, innate immunity is not as important.
because of the emergence of adaptive immunity. This supports the concept raised at the start of this chapter, that once memory is established, innate immunity is not as important.
The second TIR domain adapter to be found was Mal (also termed TIRAP).53,54 This protein is required for MyD88-dependent signaling specifically by TLR2 and especially TLR4. It appears to act as a bridging adapter for MyD88 to be recruited to the receptor TIR domains and has a PIP2 binding domain which localizes it to the plasma membrane.55 It is also a substrate for IRAK-1 and IRAK-4, and upon phosphorylation undergoes ubiquitination and degradation,56 providing a negative feedback loop in TLR signaling.
The MyD88-independent signalling pathway involves the third adapter, Trif.57 This adapter is used by TLR3 to signal and activates NF-κB as well as another transcription factor IRF3 via activation of the kinase TBK-1.58 TLR4 can also activate Trif, but this happens in endosomes. The fourth adapter, Tram, mediates this effect, acting as a bridge for Trif in a manner similar to Mal acting as a bridge for MyD88.59 Tram has a myristoylation motif that localizes it to the plasma membrane.60 However, upon activation of TLR4, Tram undergoes phosphorylation by protein kinase C-epsilon.61 This leads to dissociation of Tram from the plasma membrane, and it is likely that a TLR/Tram complex localizes to the endosome, where Trif is then recruited, again leading to TBK-1 activation and induction of type I interferons. The TLR4 signal is terminated by a splice variant of Tram termed TAG, which displaces the Trif/Tram complex.62 It is this dual pathway, Mal/MyD88 and Tram/Trif, occurring at different subcellular localizations, that probably make LPS such a powerful innate immune activator, as a wide range of genes can be induced. Figure 15.4 illustrates the TLR4 signaling pathway.
The final TIR domain adaptor to be found is SARM. This is a large protein with a TIR domain and SAM and ARM domain. SARM is a negative regulator of Trif-dependent signaling, acting as another modulator of TLR4 action.63 It is of interest that there are several mechanisms to inhibit TLR signaling, including microRNAs, decoy receptors, ubiquitin regulators (eg, A20), and splice variants of MyD88 and IRAK2 that are inhibitory.64 This tells us that TLRs, and in particular TLR4, must be kept under control otherwise the host can be damaged or even succumb.
Viruses have also been shown to target TLRs. The best examples are two vaccinia virus proteins A46R and A52R.65 A46R has been shown to block recruitment of TIR domain-containing adapters, particularly Mal and Tram.66 A52R inhibits activation of the IRAKs.67 Deletion of A52R leads to attenuation of the virus, confirming its importance for viral evasion.68
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


