Innate recognition plays an important role in determining the outcome of the host-parasite encounter by both providing an initial barrier to infection and by influencing the magnitude and class of the subsequent adaptive immune response. At the same time, from the parasite’s point of view, innate immune defenses must be subverted for infection to be established; it is clear that many parasitic pathogens have evolved specific mechanisms for evading them, and these evasion mechanisms can also provide an explanation for virulence differences amongst parasite strains. Moreover, in some cases, parasites appear to actually hijack the process of innate recognition to deviate adaptive immunity to facilitate their own persistence.
Humoral Mechanisms
Innate resistance against parasitic infection is mediated in part by preexisting, soluble factors that recognize and destroy invading developmental stages or target them for killing by effector cells. The alternative pathway of complement activation provides a first line of defense against extracellular parasites and because of this, the infective stages of parasitic protozoa and helminths have developed a various strategies to subvert complement-mediated attack. In some instances, blood and tissue parasites have evolved redundant mechanisms to ensure their survival
during serum exposure. For example, infective metacyclic and bloodstream trypomastigotes of
Trypanosoma cruzi express multiple stage-specific surface glycoproteins, such as gp160 and the 87-93kDa trypomastigote decay accelerating factor, which are actively released by the parasite and are functional homologues of human decay accelerating factor that interferes with assembly of C3 convertases by binding to C3b.
3 Another trypomastigote glycoprotein, gp58/gp68, inhibits alternative pathway C3 convertase assembly by binding to factor B.
T. cruzi trypomastigotes have also been found to continuously shed acceptor molecules with covalently bound C3 fragments, thought to be due to an endogenous phospholipase that cleaves glycosylphosphatidylinositol (GPI)-anchored membrane proteins. In addition, trypomastigotes export calreticulin to the parasite surface where, by binding C1, the protein can both inhibit activation of the classical pathway and promote parasite invasion.
4 T. cruzi amastigotes, on the other hand, have been shown to resist complement lysis by preventing membrane insertion of the membrane attack complex (MAC), C5b-9. An analogous mechanism of resistance has been observed for the infective metacyclic promastigote stage of
Leishmania, which expresses an elongated form of the major surface and released glycolipid on
Leishmania promastigotes, lipophosphoglycan (LPG), such that it behaves as an effective barrier to membrane insertion and pore formation by MAC.
5 Metacyclics also increase expression of the surface metalloproteinase gp63,
6 which can cleave C3b to the inactive iC3b form, thus preventing deposition of MAC.
7 Both C3b and iC3b effectively opsonize the complement resistant forms for uptake by macrophages, its host cell of choice. Tissue-invasive strains of
Entamoeba histolytica also activate the alternative complement pathway but are resistant to lysis due to the action of a Gal/GalNAc lectin, which mediates adherence of trophozoites to host cells and binds to C8 and C9 terminal components.
8 Interestingly, the lectin shares sequence similarities with CD59, a membrane inhibitor of MAC in human blood cells.
The damage caused to worms as a consequence of alternative pathway activation is due primarily to the bound C3 activation products that act as ligands for cellular adherence and killing by eosinophils, neutrophils, and macrophages. In addition to synthesizing their own complement regulatory proteins to subdue the activation cascade, helminths also acquire endogenous regulatory molecules from the host. For example, schistosomes can inhibit complement activation through surface-expressed parasite proteins that bind C2, C3, C8, and C9 but also do so by acquiring decay accelerating factor from the host and incorporating it into their teguments.
9 Similarly, the infective L3 stage larvae of
Onchocerca volvulus, the causative agent of river blindness, were shown to bind the main human fluid phase regulator factor H, thereby promoting C3b inactivation.
10 Other parasitic nematodes such as
Toxocara canis,
Brugia malayi, and
Trichinella spiralis appear to block complement attack by secreting proteases that attack the complement pathway
11 or regulatory proteins that inhibit its function.
12A well-characterized set of soluble mediators providing a barrier to parasitic infection are the primate-specific trypanosome lysis factors (TLF1 and TLF2) present in serum that contribute to the innate resistance of humans to
Trypanosoma brucei infection.
13 The key active components of these serum complexes are haptoglobin-related protein and apoliprotein L (ApoL)-1 that together are cytotoxic to
T. b. brucei and act synergistically to provide enhanced trypanosome killing when assembled into the same high-density lipoprotein (HDL) particle. Haptoglobin-related protein and ApoL-1 have different proposed activities; ApoL-1 is able to form ion pores in lysosomal membranes, whereas haptoglobin-related protein is able to accelerate lysosomal membrane peroxidation.
Whereas the TLFs are capable of killing
T. brucei, the species that infect humans,
T. b. gambiense and
T. b. rhodesiense, are both refractory to TLF-mediated cytolysis. This property has been correlated with the expression of a serum resistance-associated gene that is homologous to the variant surface glycoprotein. Importantly, transfection of serum resistance -associated gene from
T. b. rhodesiense into
T. brucei confers resistance to lysis by human serum, arguing that its expression may have been a critical step in the adaptation of the former parasite for infection of primates.
14 Interestingly, mice cotransfected with TLF components display enhanced resistance to
T. brucei infection, suggesting that such a genetic modification strategy might be useful in protecting livestock against this parasite, which still is a major impediment to cattle farming in many parts of Africa.
15
Cellular Mechanisms
Phagocytosis by macrophages represents an innate first line of defense against protozoan pathogens. Macrophages possess primary defense mechanisms, including activation of oxidative metabolism, which are induced by the attachment and engulfment of microbial agents, and the early survival of intracellular parasites will depend on their ability to avoid or withstand oxidative stress conditions. The major source of reactive oxygen species is the multimeric enzyme complex, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Early studies suggested that
Leishmania parasites avoid triggering the oxidative burst by actively inhibiting macrophage (PKC) activation,
16 which is required for phosphorylation of several sites on the cytosolic oxidase subunit, p47phox. The inhibition of the respiratory burst has also been linked to leishmanial LPG, which is rapidly transferred to the inner leaflet of the phagosomal membrane and prevents translocation of the NADPH oxidase cytosolic components.
17,18 As some LPG-deficient
Leishmania strains still manage to survive in macrophages, it is clear that redundant mechanisms exist for the parasite to avoid macrophage triggering. These include opsonic ingestion through receptors that are uncoupled from the activation of NADPH oxidase. A number of “silent” entry receptors have been described that are variably used by different species and developmental stages of
Leishmania, including complement and mannose receptors,
19 and receptors for apoptotic cells.
20,21In contrast to
Leishmania,
Toxoplasma enters all nucleated cells, including macrophages, by an active invasion mechanism that excludes most host cell proteins, including membrane components of the NADPH oxidase, from the
parasitophorous vacuole.
22 Malaria parasites are also sensitive to oxidative stress, and knockout mice lacking NADPH oxidase suffer more rapid increases in malaria parasite densities than wild-type mice.
23 In this case, reactive oxygen species are generated primarily as a result of the degradation of host hemoglobin within parasitized red blood cells (RBCs).
24 The detoxification of reactive oxygen species is achieved with a range of low-molecular weight antioxidants, including the tripeptide glutathione, and a number of host- and parasite-encoded enzymes.
25The maturation of phagosomes into digestive organelles represents the heart of the defensive machinery of macrophages, and intracellular parasites have evolved diverse strategies to avoid, escape from, or withstand the acidified, hydrolytic environment of phagolysosomes. For
Toxoplasma, the integral membrane proteins that are excluded from the nascent vacuole include those involved in acidification and fusion with the endosomal network.
26 If instead the parasite is forced to enter the cell by a phagocytic pathway, as a consequence of, for example, antibody opsonization, it is targeted through the normal phagolysosomal system and is killed.
27 For
T. cruzi, which trigger a wound repair pathway involving lysosome exocytosis to enter into cells,
28 the early vacuole is acidified and potentially fusogenic. Intracellular survival of
T. cruzi is dependent on its ability to escape from the vacuole, a process facilitated by its expression of a putative pore-forming protein that is immunologically cross-reactive with human C9, and that can disrupt the phagosome membrane allowing egress of the parasite into the cytoplasm.
29 Leishmania promastigotes, again via transfer of their surface LPG which increases the periphagosomal accumulation of F-actin and disrupts phagosome microdomains, transiently inhibit normal phagosome maturation.
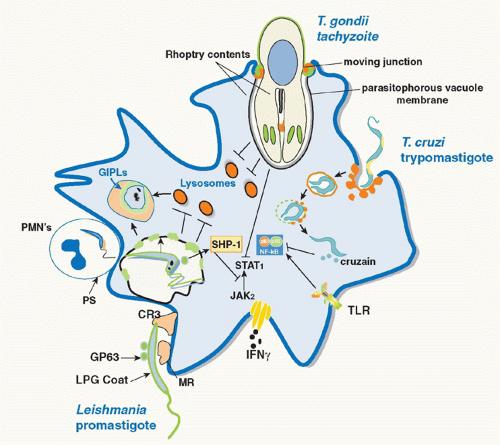
30 The delay in phagosome maturation may be necessary to allow sufficient time for metacyclic promastigotes transmitted by the sand fly to differentiate into more acidophilic, hydrolase-resistant amastigotes. The various strategies employed by parasitic protozoa to evade the innate defenses of host macrophages are depicted in
Figure 38.1.
Neutrophils have been an understudied component of the innate cellular response to protozoan pathogens, despite the fact that they are rapidly and massively recruited to the site of parasite delivery by the bites of arthropod vectors. They have been clearly revealed by intravital two-photon microscopy to be the first cells to take up
Leishmania in the skin during the first hours of infection following inoculation by needle or vector sand flies.
31,32 Their localized recruitment is triggered by the vascular damage caused by the needle injection or the sand fly bite in addition to signals derived from sand fly saliva and the parasite.
33,34 The survival of
Leishmania following their phagocytosis by neutrophils, similarly to macrophages, is dependent on their ability to inhibit fusion of tertiary granules with the parasite-containing phagosome, which was again shown to be linked to the expression of the promastigote surface LPG.
35,36 Interestingly, macrophages have been observed to phagocytose infected neutrophils in vitro,
35,37 and the exploitation of the apoptotic cell clearance function of macrophages is the basis for the “Trojan horse” infection model whereby infected, apoptotic neutrophils are proposed to silently deliver
Leishmania to host macrophages to initiate productive infections in these cells. At later stages of infection, neutrophils may help to defend against
Leishmania, in part, by releasing fibrous deoxyribonucleic acid (DNA)-based extracellular traps that ensnare and kill the parasite.
38 DNA-based extracellular traps are also elicited by
T. gondii tachyzoites, with evidence that they exert direct microbicidal effects and interfere with host cell invasion and parasite spread.
39 Although neutrophils are not required to control malaria infections (and may, indeed, contribute to tissue damage), their oxidative burst is severely compromised following exposure to the heme detoxifying enzyme heme oxygenase, leaving malaria patients at increased risk for systemic, gram-negative, bacterial infections.
40Unlike protozoa, helminths are too big to be engulfed by phagocytes and can only be killed by these cells when the latter have been activated by products of the adaptive immune response. Instead, eosinophils, which frequently accumulate in tissues soon after worm invasion, may mediate innate cellular defense against helminth larvae by means of discharge of the major basic protein and cationic proteins present in the granules of these cells.
41,42In contrast to intracellular killing by phagocytes and extracellular killing by eosinophils, some innate cellular defenses do not eliminate parasites directly but instead trigger other effector cells to do so. Perhaps the best studied example of this form of innate immunity is the natural killer (NK) cell pathway of cytokine production. NK cells become activated as a consequence of various parasitic infections;
Leishmania promastigotes;
Plasmodium falciparum -infected RBCs; components of
T. gondii,
T. cruzi,
E. histolytica, and
Cryptosporidium parvum; and excretory-secretory proteins of the hookworm
Necator americanus43 all activate human peripheral blood NK cells to produce interferon (IFN)γ. Despite occasional reports of direct binding of parasite ligands to NK-cell receptors, the emerging consensus is that NK-cell activation is secondary to pattern recognition receptor (PRR)-mediated activation of myeloid dendritic cells (DCs) and monocyte/macrophages and requires both contact-dependent and cytokine-mediated (interleukin [IL]-12, IL-18) signals.
44 Moreover, in the absence of Th2 responses, NK cells may become an important source of the protective type-2 cytokine, IL-13, during murine gastrointestinal nematode infections.
45 These findings suggest that NK cells may provide a T-lymphocyte independent pathway for cytokinemediated defense and as such serve to prevent parasites from overwhelming the host prior to the development of adaptive responses. Nevertheless, there is increasing evidence that NK responses are markedly enhanced by T-cell-derived IL-2, revealing a novel pathway by which adaptive immune responses may augment innate responses.
46,47 Trafficking of NK cells to parasite-infected tissues is critically dependent upon chemokines binding to CCR5.
48 Both IL-10 and transforming growth factor (TGF)-β have been shown to serve as negative regulators of NK cell IFNγ production by means of their suppression of monokine and B7 expression by antigen-presenting cells (APCs) or, in the case of TGF-β by directly affecting NK-cell function.
49 Such suppression
may be important in protecting the host against the tissue damaging effects of excessive NK-cell-derived IFNγ and tumor necrosis factor (TNF)-α. NK-cell responses are further regulated by calibration of signals from activating and inhibitory receptors for major histocompatibility complex (MHC) molecules; moderation of NK responses to malariainfected RBCs by inhibitory receptors such as NKG2A/CD94 and polymorphic killer-cell Ig-like receptors has been proposed.
50 Although NK-cell-derived IFNγ can limit the initial phase of protozoal replication
51 and may a play a role in the polarization and expansion of Th1 cells, in some situations, adaptive T-cell immunity is sufficient to control infection even in the absence of this early NK response.
52,53 The role of NK-cell cytotoxicity in resistance to protozoan infection is less well understood; for murine malaria, it is not required for NK-mediated resistance to blood stages,
52 but cytotoxic NK killing of malaria-infected liver cells has been reported.
54Two other cell populations that may function to provide a rapid cytokine response to invading parasites are γδT cells and NK T cells. These “unconventional T lymphocytes” express T-cell receptor chains of limited diversity, which may be designed for innate recognition of microbial structures or self-components revealed by infection of host cells. Although the function of NK T cells in innate resistance to parasites is currently under debate, there is considerable evidence supporting a protective role for γδT cells. Although representing a small percentage of lymphocytes in the periphery, γδT cells are abundant in epithelial and mucosal tissues, the sites of initial host invasion by many parasites. Moreover, their numbers increase in peripheral blood in response to a number of protozoan infections
55 where they can contribute effector cytokines or, in the case of extracellular
P. falciparum merozoites, mediate direct granulysin-dependent killing.
56 Nevertheless, rather than being essential for host resistance, it is likely that γδT lymphocytes (in common with NK cells) provide an adjunct to conventional αβ CD4+ and CD8+ T cells in restricting parasite growth during the vulnerable period when the adaptive responses mediated by these lymphocyte subsets is emerging.
57,58A recently discovered group of effectors in the early cellular response to parasites are the innate lymphoid cells (ILCs). This cell population belongs to a heterogeneous family of innate non-T, non-B cells that are not antigen restricted. However, as they express CD45 and are dependent on traditional T-cell growth factor signaling pathways, they have been called ILCs. In common with NK cells, which themselves have now been reclassified as ILC, ILC2 produce cytokines important in T-cell subset differentiation and amplification. In the case of ILC2, these cytokines are IL-4 and IL-13, which promote Th2 development and function. As outlined later in this chapter, ILC2 (which may comprise as many as four distinct cell populations) play an important role as a major source of IL-13 in the intestinal response to nematodes and in worm expulsion.
59 Also as discussed subsequently, basophils can provide an innate source of IL-4, thereby driving Th2 differentiation, and have been proposed to do so while simultaneously serving as APCs for T-cell activation.
Role of Pattern Recognition Receptors in Innate Recognition of Parasites
The innate immune system, in addition to providing a natural barrier that limits infection, also plays a critical role in the initial recognition of parasites and the triggering of adaptive immunity. Invading parasites, such as other pathogens, are sensed by host PRRs that recognize microbe-associated molecular patterns shared by different groups of organisms
(Table 38.2). These PRRs are highly expressed
on both epithelial cells and APCs and, when ligated, trigger cytokine and costimulatory signals that initiate both innate and adaptive cellular responses.
Toll-like receptors (TLR) are, so far, the major group of PRRs known to be triggered by parasites. The study of TLR involvement in parasitic infection began with the identification of parasite ligands that stimulate cytokine production from macrophages and DCs. In the case of protozoa, important classes of such ligands are the GPI lipid anchors present on many parasite surface proteins and phosphoglycans, and the membrane-associated glycoinositolphospholipids. Thus, GPIs from
T. brucei,
Leishmania,
T. gondii, and
P. falciparum can stimulate macrophages to upregulate inducible nitrogen oxide synthase expression and produce proinflammatory cytokines. Similarly, the GPI anchor fraction of mucin-like molecules from
T. cruzi trypomastigotes triggers macrophage production of IL-12 and TNF. Glycoinositolphospholipids from
T. cruzi and
T. brucei possess similar agonist activities.
60 Studies employing reporter cell lines transfected with specific TLRs, or TLR-specific knockouts, have demonstrated that the responses induced by these parasite glycolipids are due to stimulation of TLR2 and to a lesser extent TLR4 (preferentially triggered by glycoinositolphospholipids in the case of
T. cruzi).
Parasite nucleic acids also represent important ligands for TLR recognition. Thus, genomic DNAs from several protozoan species have been shown to stimulate host proinflammatory cytokine production presumably through the recognition of unmethylated CpG motifs by TLR9. A major role for nucleic acids in stimulation of host resistance to
T. cruzi is evidenced by the diminished host resistance of both TLR9 and TLR2/TLR9 double knockout animals
61 and by more recent studies on mice deficient in UNC93B1, a protein that mediates translocation of TLR3, 7, and 9 to endolysosomes. These animals showed greater susceptibility to infection than TLR9-deficient mice, a finding that points to an additional role for TLR7 recognition of parasite RNA in the innate response to
T. cruzi.62 TLR9 signaling also contributes to host resistance to
Leishmania apparently through DNAd-riven activation of both DCs and NK cells, although there is debate as to whether this stimulation also plays a role in Th1 response development.
63,64 Leishmania strains isolated from patients with mucocutaneous leishmaniasis were found to harbor high amounts of a ribonucleic acid virus that induced a TLR3-mediated hyperinflammatory response in mice that may explain the destructive metastatic lesions associated with mucosal disease.
65 In the case of
P. falciparum, hemazoin, a product of malaria-induced hemoglobin degradation is a TLR9 agonist,
66 although at present there is controversy as to whether this results from contamination with immunostimulatory DNA fragments of parasite origin.
67A chemically different class of TLR ligands are the profilin proteins expressed by apicomplexan protozoa. These molecules are unique to eukaryotes and are typically associated with intracellular actin. Profilins from
T. gondii,
Eimeria, and
C. parvum potently trigger IL-12 production from murine DCs as well as systemically following in vivo inoculation.
68 Experiments in the murine
T. gondii model established that this response is due to the stimulation of TLR11, a TLR that although present in mice and other small animals is not functionally expressed in primates. Recent studies have demonstrated that UNC93B1 is required for triggering of TLR11 activation and IL-12 production by
T. gondii profilin in mice, thus revealing an endosomal localization for this TLR.
69 Nevertheless, whereas UNC93B1-deficient mice are highly susceptible to
T. gondii infection, animals deficient in TLR11
68 (as well as TLR3, 7, and 9) show no or only partial impairment of resistance, indicating that UNC93B1 must serve additional functions in innate immunity to the parasite beyond its role in TLR11 localization and activation.
70Helminth parasites also express TLR ligands, although as discussed subsequently, their role in the immune response has been harder to define than those characterized in protozoa. Moreover, in many studies, the possibility of contamination by bacterial or viral TLR ligands from symbionts has not been systematically ruled out. Indeed,
Wolbachia symbionts confer strong TLR4 and TLR2 agonist activity on filarial parasites due to their endotoxin-like components.
71 Well-studied examples of helminth TLR microbe-associated molecular patterns include the phosphorylcholine containing moieties of the filarial glycoprotein ES-62
72 and the lysophosphatidylserine components of schistosome membranes
73 that trigger TLR4 and TLR2, respectively. In addition, schistosome eggs possess double stranded ribonucleic acid molecules that stimulate TLR3 in DCs.
74The role played by TLRs in the host response to parasites is complex and not fully delineated in any of the host-parasite models studied. The main evidence for TLR involvement comes from experiments in mice deficient in myeloid differentiation primary response gene 88 (MyD88), a major adaptor protein required for signaling by most TLRs as well as by the IL-1 and IL-18 receptors. MyD88-deficient animals exhibit a loss in resistance to
T. gondii,
T. cruzi,
L. major,
T. brucei, and—in the case of
Plasmodium berghei—decreased immunopathology
60 that likely reflects the role of TLR signaling in accessory cells (eg, DCs, epithelial cells) in the initiation and maintenance of Th1 responses. However, the altered susceptibility of these mice could also involve non-TLR-related effects of MyD88 deficiency and/or T-cell intrinsic functions of the gene, as demonstrated in the
T. gondii mouse model.
75 Significantly, no major alterations in helminth-induced immune responses have as yet been described in MyD88- deficient hosts on susceptible genetic backgrounds, arguing against a significant role for TLR signaling in Th2-dependent host resistance and pathology in worm infections.
In contrast to the dramatically increased susceptibility often observed in MyD88-/- mice following protozoan infection, mice deficient in single TLRs exposed to the same parasites rarely show pronounced changes in resistance, even when such animals display major immune response impairments. For example, while TLR11-/- mice infected with
T. gondii develop severely blunted IL-12 responses both in vitro and in vivo, they nevertheless survive the acute-phase infection and show only a minor increase in parasite load in comparison to fully susceptible IL-12-deficient animals.
68 Such findings may reflect redundant functions for different TLR or a requirement for multiple MyD88-dependent TLR (or IL-1/IL-18) signals in host resistance. An additional
complexity is that mice deficient in single TLRs may show unaltered (or even enhanced) resistance because the TLR in question controls an immunopathologic response or downregulates host effector functions.
76 Moreover, as exemplified by recent studies in the peroral
T. gondii infection model, TLR signals triggered by host commensal flora (presumably as a result of intestinal barrier breakdown) can also influence the outcome of parasitic infection.
77 A major challenge of current research in this area is to decipher such positive and negative TLR signals triggered by parasite, commensal, or host ligands and establish how their integration governs host resistance.
There is also a dearth of knowledge concerning the function of PRR signaling pathways outside the TLR family. A topic of particular interest in this regard is the role of inflammasome activation in parasite-induced immunopathology. Two recent studies have demonstrated that malaria hemazoin is a potent activator of the NLRP3 inflammasome in vitro but have yielded conflicting results on the involvement of this pathway in cerebral malaria in vivo.
78,79 Most recently, sensing of AT-rich malarial DNA by an an unknown receptor that signals via the stimulator of IFN gene (STING), TANK-binding kinase 1 (TBK1), and interferon regulatory factor (IRF)3 to IRF7 signaling pathway has emerged as an entirely novel pathway of pattern recognition.
80DCs play a major role in linking parasite pattern recognition signals to the induction of both NK-mediated innate responses and T-cell-dependent adaptive immunity. As discussed previously, in the case of many protozoa, DC-derived IL-12 provides a major stimulus for the initiation of host defense pathways. The critical role of DCs is underscored by the impaired IL-12 production as well as impaired control of protozoan (
T. gondii) infection
81 in mice in which CD11c+ DC populations have been genetically depleted. Such DC-depleted mice are also unable to generate CD8+ cytotoxic T-lymphocyte responses against
Plasmodium yoelli sporozites.
82 The requirement for protozoan invasion in DC activation is complex and depends on the parasite species and DC subset in question. Indeed, as discussed subsequently, infection of DCs can result in suppressed responsiveness to activation stimuli. Nevertheless, infection of DCs with, for example, live
Leishmania83 or
T. gondii appears to be important for efficient priming of CD8+ T cells despite the sequestration of many of the protozoa in question within parasitophorous vacuoles physically removed from the class I MHC antigen presentation machinery of the host cell.
84 In addition, it appears that under certain situations (eg, immunization with irradiated malarial sporozoites
85) infected, apoptotic host cells are taken up by DCs as a mechanism of CD8+ T-cell priming. Thus, although live infection is clearly important for efficient T-cell response induction, direct infection of DCs (as opposed to other host cells) may not be required in vivo. The failure to observe parasite-containing DCs engaged in long-lived contact with T cells in in vivo imaging studies lends credence to this hypothesis but is subject to alternative interpretations.
86,87In addition to their role in initiating immune responses, DCs appear to be efficient vehicles for dissemination of parasites into different tissues as illustrated by studies in the
T. gondii mouse model.
88 Infection of DCs can also serve as a survival strategy for protozoa and as a means for maintaining cryptic infection, as suggested by recent studies in which DCs from mice with rodent malaria were found to be capable of initiating infection when inoculated into naïve hosts.
89
Downregulation of Innate Signaling Pathways by Parasites: Role in Virulence Determination
In addition to upregulating APC function, parasite products can also dampen their activity either as a mechanism of immune evasion or for the purpose of protecting the host against the pathology associated with an uncontrolled immune response.
Leishmania,
T. cruzi, and
Toxoplasma have in common their ability to inhibit proinflammatory and IFNγ inducible responses in infected macrophages, such that the parasites might not only prevent or delay the induction of Th1 responses but also render infected macrophages unresponsive to activation signals during the effector phase of the immune response (see
Fig. 38.1). In many cases, the parasites exploit the presence of host cell phosphatases whose normal role is to temper the magnitude and duration of the proinflammatory response. For
Leishmania, the inhibition is due in part to activation by the
Leishmania metalloproteinase gp63 of host cell protein tyrosine phosphatases, including SHP-1 and other nonreceptor protein tyrosine phosphatases, which inactivate Janus kinase (JAK) family members involved in the IFNγ inducible phosphorylation cascade.
90 Other host cell phosphatases were found to be activated by
Leishmania cysteine proteinases to regulate the Mitogen-activated protein kinase (MAPK) family members p38 and extracellular signal-regulated kinase (Erk)1/2, resulting in the upregulation of IL-10 and downregulation of nitrogen oxide (NO) and TNF-α production.
91 T. cruzi trypomastigotes, via expression of GPI-anchored mucin-like molecules, also activate macrophage phosphatases that target downstream elements of the TLR pathway, including MAPK and nuclear factor-kappaB (NF-κB), to establish a state of tolerance in the infected cells.
92 Furthermore, NF-κB p65 was found to be targeted directly for cleavage by the
T. cruzi protease cruzain, and cruzain-deficient parasites rapidly activated macrophages via NF-κB p65 for IL-12 expression.
93 A homologue of cruzain, cysteine peptidase B, is expressed in
Leishmania mexicana and was likewise found to degrade NF-κB p65 to inhibit IL-12 production by infected macrophages.
94 The need to counterbalance the excessive inflammation that can be triggered by
T. gondii infection extends to the modulation of IFNγ-induced responses, and in particular the signal transducer and activator of transcription (STAT)1 signaling cascade that is critical for resistance to
T. gondii. Various defects in IFNγ-initiated STAT1 signaling have been described in
T. gondii-infected cells, including proteolytic cleavage of STAT1, dephosphorylation, and prevention of nuclear translocation.
95 Thus, although both
T. cruzi and
T. gondii initiate strong proinflammatory responses in host macrophages, these signaling pathways appear to be subsequently impaired to avoid reaching pathologic levels that may be detrimental to the host and/or lethal to the parasite during the adaptive phase of the immune response.
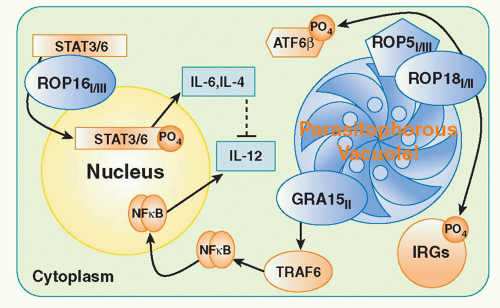
The differential ability of parasites to dampen cellular activation signals appears to be a major factor in virulence
determination. This has been elegantly studied in recent work on
T. gondii examining the basis of the marked virulence differences in mice between Type I, Type II, and Type III strains of the parasite. By examining the progeny of genetic crosses between Type I and III or Type II and III parasites, two highly polymorphic genes ROP18 and ROP16 were identified as major virulence determinants.
96,97 Both genes encode serine-threonine kinases associated with a parasite organelle known as the rhoptry. During the process of invasion, rhoptry proteins are discharged into the host cell. Studies with ROP18 indicate that in Type I strains the kinase inactivates effectors known as immunity-related GTPase (IRG) proteins (discussed subsquently) that mediate parasite killing by disrupting the parasitophorous vacuole.
98 By contrast, the Type 1 variant of ROP16 transits to the host cell nucleus where it phosphorylates and activates STAT3 and STAT6, which in turn downregulate the proinflammatory IL-12, IL-6, and inducible NO synthase response to the parasite via their roles as positive regulators of silencers of cytokine signaling and/or IL-10.
96 Recently, a third virulence factor has been identified, in part, through a new genetic cross between Types II and I. ROP5 is a pseudokinase and serves as a scaffold to enable ROP18 activity.
99,100 The mechanism(s) by which these three rhoptry proteins interact in suppressing host cellular function is a major area of interest in the field. A current model of the function of the major
T. gondii virulence determinants is presented in
Figure 38.2.
In addition to their suppressive effects on macrophages and other host cells, parasites or their products can negatively regulate DC function, both as a means to delay the onset of the adaptive response or to restrain an ongoing response to prevent immunopathology. For example, IL-12 production by splenic DCs is rapidly suppressed following initial in vivo stimulation with soluble
T. gondii antigen and cannot be restimulated for approximately 1 week thereafter. The antiinflammatory outcome appears to result from the induction, by parasite products, of lipoxin A4, an arachadonic acid metabolite that upregulates silencers of cytokine signaling-2 to inhibit soluble
T. gondii antigen-induced DC migration and IL-12 production.
101 Similarly, in murine malaria, the initial burst of proinflammatory cytokines is down regulated as DCs become refractory to further TLR signaling.
102 The ligands implicated in these interactions include a conserved domain of
P. falciparum erythrocyte membrane antigen and hemozoin that has been shown to directly inhibit the maturation of human DCs.
103 This TLR tolerance is similar to that induced by endotoxin and may explain the prevalence of “regulatory” CD11c
lowCD45RB
high DCs later in infection that have been shown to induce IL-10-secreting T cells as a negative feedback mechanism to control immunopathology.
104 The relevance of these findings to human disease is supported by the observation that the interethnic differences in malaria infection outcome in Malian children are associated with reduced expression of activation markers and reduced TLR-induced responses in DCs from
P. falciparum-infected Dogon children who experience more severe disease.
105 Also, the percentage of human leukocyte antigen-DR + DCs has been reported to be significantly lower in Kenyan children with severe or mild malaria compared to healthy controls, and they also had increased frequencies of DCs expressing BDCA-3, a marker that is upregulated on IL-10 treated monocyte-derived dendritic cells (MDDC).
106 In Chagas disease, compromised DC function has been linked to immune suppression in chronically infected mice.
T. cruzi blood stages inhibited the lipopolysaccharide (LPS)-induced activation of mouse bone marrow-derived DCs, with both IL-10 and TGF-β important in the induction of the regulatory DC phenotype.
107,108 Together, these studies involving
T. gondii, malaria, and
T. cruzi suggest that parasites drive DC activation and proinflammatory reactions during the acute stages of infection, followed by the emergence of regulatory DCs that modulate the adaptive response, limiting both immunopathology and pathogen clearance.
By comparison, studies with
Leishmania suggest that for most strains their initial encounters with DCs fail to activate these cells and, as a consequence, proinflammatory responses and cell-mediated immune mechanisms are effectively inhibited or delayed even during the acute stage of infection. In the case of
Leishmania major, the infective
promastigotes deposited in the skin by vector sand flies were found to be poorly taken up by DCs in vitro. However, later in infection, efficient uptake of amastigotes by DC in vitro and in vivo is dependent on parasite-reactive IgG binding to FcγRI and FcγRIII; this primes DCs for efficient production of IL-12.
109 Furthermore, the initial encounter of other
Leishmania species associated with nonhealing infections in mice not only failed to activate DC but also inhibited their subsequent responses to activation signals.
110 For example, DCs with clear regulatory properties and bearing the phenotype of CD11c
lo CD45RB+ CD11b+ IL-10-producing cells, emerge as the predominant DC subset in the spleen of
Leishmania donovani-infected mice and induce antigenspecific tolerance in vivo.
111
Mechanisms Underlying Th1/Th2 Response Selection
Because parasites often stimulate CD4+ T-cell responses that are highly polarized in either the Th1 or Th2 direction, parasitic infection models have become important tools for studying the cellular basis of Th1/Th2 response selection. DCs are thought to be an important source of the signals that determine CD4+ T-cell effector choice, and their role is best understood for Th1 responses.
T. gondii,
T. cruzi, and
Plasmodium have been shown upon their initial encounter with DCs to upregulate expression of IL-12 and costimulatory molecules. The nature of these encounters has been extensively studied for
T. gondii, whose possession of important Th1-inducing TLR ligand(s) has been inferred by the high susceptibility to infection of mice lacking MyD88
112 and shown to be selectively acting on DCs.
93 Importantly, the high susceptibility of MyD88-/- mice is comparable to that observed in IL-12-/- mice and is not due to the absence of IL-1/IL-18 signaling.
113 As noted previously, the stimulation of TLR11 by parasite profilin appears to be the major MyD88 pathway that triggers this IL-12 response in the murine model.
56 T. cruzi induces a delayed, although ultimately strongly and persistently polarized, Th1 response in infected mice that is also MyD88 dependent and, as already mentioned, appears to be induced largely by nucleic acid ligands.
91 The Th1 responses that contribute to immunopathology during blood-stage malaria infections are driven, at least in part, by TLR ligands as MyD88-/- mice have decreased production of IL-12 and attenuated pathology,
114 whereas—in acutely infected mice and humans—hyperresponsiveness has been linked to IFNγ-induced enhancement of TLR expression on DCs.
115 As previously discussed, hemozoin or a hemozoin-DNA complex hemozoin acting through TLR9, and GPI binding to TLR2, appear to be the main malaria component(s) that activate mouse and human blood DCs to secrete IL-12.
116,117,118Unlike the protozoan pathogens just described,
Leishmania appears to trigger TLR signaling in DC poorly, and in most cases, their ability to activate these APCs—for upregulated expression of costimulatory molecules and especially for IL-12 production—requires additional signals that are host derived. For example, the interaction of CD40L on T cells with CD40 on infected DCs enhances IL-12p70 secretion in vitro
119 and is essential for
L. major -specific Th1 activation and immunity in vivo.
120 In addition to IL-12, there are signals delivered by other cytokines, including IL- 18, IL-27, IFN α, and IL-1, which have been shown to bias
Leishmania-specific CD4+ T-cell priming toward a Th1 cell fate. Mouse strains with intrinsic deficiencies in host factors necessary to augment Th1-polarized responses against
Leishmania are highly susceptible to infection, and as noted previously, the first direct demonstrations of the relevance of the Th1/Th2 balance to the regulation of disease outcome in vivo were based on studies of
L. major infection outcome in different inbred mouse strains. The Th2 polarization that determines the extreme susceptibility of BALB/c mice to
L. major is due, at least in part, to an intrinsically poor Th1-differentiating capacity, as even in the absence of IL-4 or IL-4 receptor signaling, IFNγ responses remain relatively weak.
121,122 An additional strain difference that may influence Th subset development relates to the finding of very rapid dissemination of parasites from the site of inoculation to the draining lymph nodes and spleen in BALB/c mice, whereas early parasite containment is observed in resistant mice.
123 These differences in parasite dissemination raise the possibility that distinct populations of DCs, with the capacity to induce preferential priming for either Th1 or Th2 cells, are present in different tissue environments and become differentially activated in resistant versus susceptible mice. In support of this hypothesis, targeting to selective DC subsets can be achieved by altering the site of antigen delivery;
L. major parasites delivered intravenously, intranasally, or even to different skin environments can elicit Th2 responses and nonhealing infections in normally resistant mice.
124,125Although L. major infection in BALB/c mice remains an extraordinary tool for investigating the factors controlling Th2 response selection in vivo, the fact that the model reflects an aberrant response arising, at least in part, from inherent Th1 developmental defects suggests that these defects might not reflect the mechanisms underlying the Th2 polarization that is a hallmark of helminth infections in virtually all mammalian hosts. Furthermore, although Th2 immune deviation is clearly an inappropriate host response to an intracellular pathogen such as Leishmania, Th2 responses are an evolutionarily driven, integral aspect of acquired resistance to most parasitic worms or to the containment of the immunopathologic reactions that worms or their products can provoke.
DCs conditioned by exposure to helminth products polarize naïve T cells toward a Th2 phenotype.
126,127 Importantly, Th2 polarization driven by most helminth antigens is MyD88 independent,
128 whereas Th1 differentiation is in most instances MyD88 dependent. Furthermore, activation of DCs by helminth antigens appears to be minimal as judged by the absence of upregulated MHC and costimulatory markers, cytokine production, and transcriptional or proteomic signatures.
129 In the case of DCs exposed to schistosome egg antigens (SEAs) or
Fasciola hepatica (liver fluke) tegumental antigen, their maturation and IL-12 p70 production in response to TLR ligands become severely impaired, associated with defective MAPK and NF-κB activation.
130,131,132 In human filariasis, live microfilariae of
B. malayi modulate DC function by altering TLR3 and TLR4 expression and function.
132 The major component of
Schistosoma mansoni eggs responsible for conditioning DCs for Th2 polarization
is a secreted T2 ribonuclease omega-1 that is hypothesized to act by limiting conjugate formation between DCs and CD4+ T cells.
133,134 N-glycans containing fucose and binding to DC-specific intercellular adhesion molecule 3-grabbing nonintegrin on DCs have also been implicated in the induction of Th2 responses by schistosomes.
135Many of these observations would seem consistent with a model of Th2 induction by DCs that, in the absence of a threshold of instructional, positive signals for Th1 priming occurs via a default pathway. However, defaulting to Th2 is not observed when IL-12-deficient mice are infected with either
T. gondii or
Mycobacterium avium.136 Furthermore, in the case of DCs exposed to SEA, there remains a requirement for NF-κB signaling and costimulation (CD40 and OX40L) for the induction of Th2 responses.
137 Therefore, it is more likely that helminth-conditioned DCs, rather than initiating a default pathway in naïve T cells, provide a set of active, instructive signals that result in Th2 priming. The exact nature of these instructive signals is, however, currently unclear.
Surprisingly, IL-4 does not appear to provide an essential instructive signal for Th2 differentiation, as helminth-conditioned DCs will polarize a Th2 response in vitro in the absence of IL-4,
126 and mice deficient in IL-4, IL-4R, or STAT 6 develop diminished but still physiologically significant Th2 responses when infected with
Nippostrongylus brasiliensis or
S. mansoni.138,139 Using T-cell-specific gene ablation of Notch1 and 2 receptors, it was revealed that Notch is required for Th2 responses to
Trichuris muris140 and also drives IL-4R/STAT 6 independent Th2 differentiation in vitro in response to SEAs.
141 Although DC expression of the Notch ligand, Jagged-2, has been shown to be required for Th2 differentiation in response to SEA in vitro, it is apparently not required for Th2 polarization induced by SEA-pulsed DCs in vivo.
142Although IL-4R/STAT 6 signaling is not essential for priming of IL-4+CD4+ T lymphocytes in many helminth infection models, it is clear that IL-4 plays a critical role in the maturation and stabilization of Th2 cells once their phenotype has been decided. In this context, Th2 cells need not be the only source of IL-4 for maturation of the Th2 response; basophils committed to express IL-4 are recruited to the liver and lungs of mice infected with
N. brasiliensis,
143 and a direct role for basophils in helminth-induced type 2 immunity was confirmed by the capacity of adoptively transferred IL-4 producing, adult-derived basophils to restore the ability of juvenile mice to expel
N. brasiliensis.
144 It has been argued that basophils rather than DCs are the critical cells responsible for helminth-induced Th2 polarization (see following discussion), but this hypothesis has been challenged and may be limited in validity to those experimental models where IL-4 appears necessary for Th2 priming.