FIGURE 144-1. Formation of the morula with production of human chorionic gonadotropin (hCG), the first signal to the mother of the presence of pregnancy. As cell division occurs, hCG production is increased. The outer cells cease to express homeobox protein NANOG, which is required for totipotency, and form the trophectoderm and later the placenta. These cells transport fluid in an inwards direction, leading to the formation of the blastocyst. The central cells which continue to express NANOG retain totipotency and form the inner cell mass of the blastocyst and later the embryo. Prior to attachment of the blastocyst to the endometrial lining of the uterus, the blastocyst hatches from the zona pellucida. The blastocyst then penetrates the decidualized endometrium, and the trophectoderm begins to invade.
Manipulating Maternal Metabolism to Transfer Nutrients to the Embryo and Regulating Growth of the Fetus
The placenta secretes a range of hormones that regulate maternal metabolism to promote transfer of nutrients to the fetus and regulate fetal growth.
Trophoblast cells differentiate into an invasive phenotype in the hypoxic environment of early pregnancy when the blastocyst lies within the tissue of the decidua. Cytotrophoblasts invade through decidual tissue until they reach the uterine spiral arteries, where changes in oxygen tension lead to the expression of genes that alter the cytotrophoblast cell phenotype towards that of endothelial cells. The modified cytotrophoblast cells replace the vascular lining and also destroy the muscular walls of the spiral arteries, changing them from high-resistance to low-resistance vessels, thus increasing the blood flow rate to the developing placenta and embryo.
Placental invasion into the spiral arteries is accompanied by other structural changes. Cytotrophoblasts sit on the maternal side of the basement membrane that lies between the maternal and fetal tissues. Some cytotrophoblasts proliferate and differentiate into columns of cells that form an attachment site to the wall of the uterus, and other cells differentiate into syncytiotrophoblasts and fuse to form the syncytium that lines the vascular cavities created by erosion of maternal spiral arteries and associated veins (Fig. 144-2). Differentiation of cytotrophoblasts into syncytiotrophoblasts is regulated by the expression of a retrovirus envelope protein called syncytin, which promotes fusion into the multinuclear syncytiotrophoblasts.5 The expression of syncytin is promoted by high levels of cAMP that likely result from the autocrine stimulation of cytotrophoblasts by hCG. Downstream signaling from the hCG receptor via Gαs proteins, adenylate cyclase, and protein kinase A promote acetylation of the transcription factor GCMa, which directly stimulates expression of syncytin.6 Thus, formation of the human placenta requires the expression of a protein derived from the evolutionary distant incorporation of a retrovirus into the human genome.
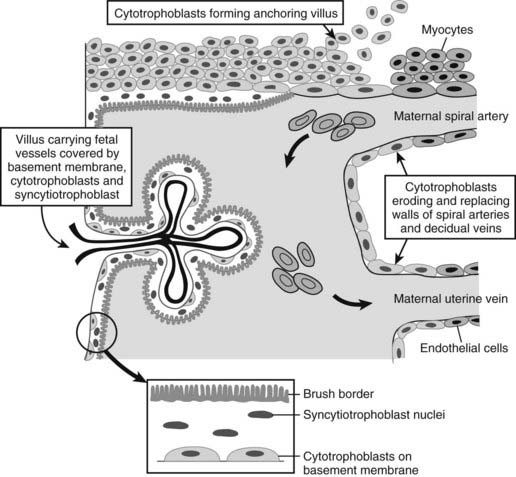
FIGURE 144-2. Cytotrophoblasts grow into the maternal decidua from a basement membrane. The cytotrophoblasts differentiate into several cell types. Some form dense columns of cells that form anchoring villi that attach the placenta to the lining of the uterus, others erode into the maternal spiral arteries and transform into an endothelial phenotype that lines these vessels, and a further group fuse to form the syncytiotrophoblast layer that lines the villus space.
The transfer of substances from the maternal to fetal circulation occurs by active transport or by passive diffusion. This process is influenced by factors such as the speed of blood flow, the size of the molecule, and the concentration gradient between the maternal and fetal circulations. Glucose is passively transported by the glucose transporter 1, a carrier molecule.7 Amino acids are actively transferred through transporters such as the System A, L, and ASC transporters,8 while fatty acids can be transferred by passive diffusion or via transport proteins.9 The placenta is also the site of gas exchange between mother and fetus and allows the transfer of hormones and peptides within certain size limitations.
Effective placental development is critical for the delivery of nutrients and regulation of fetal growth. Placental maldevelopment and malfunction is associated with fetal growth retardation and maternal disease such as preeclampsia. Loss of regulation can lead to macrosomia and birth complications. The placenta is also the source of numerous circulating hormones and factors that enter the fetal and maternal circulations, promoting fetal development and adapting maternal physiology for pregnancy. Placental products have a further autocrine role in ongoing placental formation, growth, and nutrient transfer.
THE GROWTH HORMONE/PROLACTIN SUPERFAMILY
Five genes make up the human growth hormone cluster located on chromosome 17: GHn and GHv (encoding pituitary and placental growth hormone, respectively), PL-L, PL-A, and PL-B (both the latter encoding human placental lactogen).10 Expression of GHn is specific to the pituitary, determined by three Pit-1 response elements within the locus control region. The other four genes have additional upstream “P elements” that promote expression in the syncytiotrophoblasts and extravillous cytotrophoblasts of the placenta rather than the pituitary.11 The two growth hormones have both somatogenic and lactogenic activity at the growth hormone and prolactin receptor. Human placental lactogen possesses weak GH-receptor agonist activity but is predominantly a lactogen, along with the structurally and biologically related protein, prolactin, which acts only at the prolactin receptor. These four hormones all stimulate insulin-like factor 1 (IGF-1) production. A degree of functional overlap, and thus redundancy, is apparent in their similar metabolic and growth-potentiating actions and may allow compensation for the deficiency of one hormone.12
The three PL genes are actively induced in early gestation, although PL-L has no identifiable protein product. Transcription of PL-A and PL-B results in the tonic secretion of human placental lactogen (hPL) into both maternal and fetal compartments. Regulation of hPL production by syncytiotrophoblasts is poorly understood but may involve estrogens, glucocorticoids, apolipoproteins, and high-density lipoproteins.13,14 Maternal hPL is detectable early in gestation and rises up to 6000 ng/mL in late gestation. Fetal hPL is at 1/100 of the maternal concentration15 but is believed to contribute to fetal growth and development.
Expression of GHv by the syncytiotrophoblasts results in secretion of placental growth hormone (PGH), which differs from its pituitary counterpart by 13 amino-acid residues and is unique to pregnancy. Unlike hPL, PGH is only secreted into the maternal compartment, where it becomes detectable at approximately 7 weeks’ gestation, peaks by 36 weeks, and replaces pituitary GH as the major stimulus to maternal IGF-1 production.16 The regulation of PGH production is also not well understood but may be related to nutritional modulators rather than traditional hypothalamic factors.17
Under the influence of rising estrogen levels from the placenta, maternal pituitary lactotrophs increase prolactin production, increasing in maternal serum to 200 to 300 ng/mL at term.18 There is no transplacental transfer of prolactin; the fetal pituitary develops true lactotrophs around mid-gestation, and fetal prolactin levels rise steeply from 30 weeks until term, when maternal levels are approximated. Decidual prolactin production results in very high amniotic fluid concentrations, peaking at 16 to 22 weeks’ gestation then declining until term19; these may be associated with endometrial implantation, amniotic fluid, and electrolyte balance.20,21
THE INSULIN-LIKE GROWTH FACTOR SUPERFAMILY
The insulin growth factor system is composed of three ligands: insulin, IGF-1, and IGF-2, which bind to receptors that include the insulin receptor, the type 1 insulin-like growth factor receptor (IGF1R), the mannose-6 phosphate/IGF-2 receptor, and the hybrid insulin/IGF-1 receptors. Six binding proteins (IGFBPs) further modulate the actions of the growth factors.22 In particular, IGFBP-1 regulates the action of IGFs in pregnancy by limiting their availability to their receptors. Proteases which cleave the IGFBP-1 (e.g., pregnancy-associated plasma protein-A, matrix metalloprotease) therefore have pro-mitogenic effects.23,24 Phosphorylation of IGFBP-1 increases its affinity for IGF-1, whereas reduction in phosphorylated forms in pregnancy and large amounts of nonphosphorylated IGFBP-1 in fetal serum have growth-promoting effects.25 IGFBP-3 is the major carrier of IGF-1 and IGF-2 in the circulation, forming a ternary complex with either of the IGFs and an acid-labile subunit, thus acting as a reservoir for IGFs. IGFBP-3 is regulated by GH and increases during pregnancy.26 IGF-1 and IGF-2 act predominantly through the IGF-1 receptor, although IGF-2 can also act through the A form of the insulin receptor. The IGF-2 receptor binds and degrades IGF-2, limiting its actions.27
Synthesis of maternal IGFs is modulated by hPL and growth hormone, the latter initially of pituitary origin but replaced by placental GHv as pregnancy progresses. Although there is no placental transfer of IGFs, maternal IGFs affect fetal growth by influencing placental structure and function and diverting maternal nutrients to the fetus. Fetal growth is not controlled by fetal GH, but it is directly influenced by fetal IGFs and binding proteins, in turn regulated by fetal nutrition, insulin, and hPL levels. IGF-1 and IGF-2 are both expressed during uterine life, but IGF-1 predominates after birth under the stimulus of neonatal GH.22 Fetal pituitary growth hormone may have a small impact on late third-trimester growth, as evidenced by the lower birth size of neonates with hypopituitarism or GH insensitivity.28,29
The placenta is exposed to both maternally and fetally derived IGFs and synthesizes many components of the IGF system itself.30 These have roles in trophoblastic growth, turnover, and placental exchange.
CORTICOTROPIN-RELEASING HORMONE/ADRENOCORTICOTROPIC HORMONE/CORTISOL
Marked changes in the maternal hypothalamic-pituitary-adrenal (HPA) axis occur during gestation, modulated by placentally derived factors, as reviewed in Ref. 31. Placental estrogen drives the hepatic synthesis of cortisol-binding globulin (CBG), which elevates total cortisol levels. However, free plasma, salivary, and urinary cortisol levels similarly rise, indicating altered function of the entire axis. In addition, the cytotrophoblast and syncytiotrophoblast cells of the placenta synthesize both pro-opiomelanocortin and corticotropin-releasing hormone (CRH). The pro-opiomelanocortin precursor protein is extensively processed in the placental cells, leading to the release of melanocyte-stimulating peptides but minimal adrenocorticotropic hormone (ACTH).32 Significantly, CRH levels rise 1000-fold from the 8th week of gestation to term, with the steepest incline in the last 5 weeks prior to parturition. Placentally derived CRH is principally responsible for the hyperactivity of the HPA axis in late gestation. Although CRH has widespread roles in reproductive tissues (discussed later), hypercortisolism could contribute to the changes in maternal metabolism that promote nutrient transfer. The fetus is protected from maternal hypercortisolemia by the 11β-hydroxysteroid dehydrogenase (11βHSD2) enzyme in the placenta, which converts cortisol to the inactive cortisone.33 Alterations in the expression of this enzyme may modulate fetal growth and the development of the fetal stress response, reviewed in Ref. 34.
TUMOR NECROSIS FACTOR α
The placenta synthesizes and releases tumor necrosis factor α (TNF-α) predominantly into the maternal circulation,35 and plasma TNF-α concentrations rise during late pregnancy by around 45%. In animals, TNF-α levels are increased by experimentally induced maternal stressors such as exposure to toxic chemicals, inflammatory processes, and diabetes, and TNF-α induces apoptosis and eventual embryonic death. Additionally, TNF-α can also function to protect cells from apoptosis and repair injury.36 Further evidence implicates TNF-α as a key player in the altered metabolic profile of pregnancy.35
PARATHYROID HORMONE–RELATED PEPTIDE
Parathyroid hormone–related peptide (PTHrP) is secreted by both fetal and maternal reproductive tissues, including the amnion, chorion, cytotrophoblasts, syncytiotrophoblasts, endometrium, and myometrium and has multiple roles in placental development, transplacental calcium and nutrient transport, fetal growth, and bone development.37,38 During pregnancy, PTHrP has been implicated in the increased activity of the maternal renal 1α-hydroxylase, resulting in a rise in active 1,25-dihydroxyvitamin D and subsequent increase in intestinal calcium absorption, supporting the provisioning of calcium to the fetus.39
In concert, these placental factors drive dramatic shifts in pregnancy physiology which favor the developing fetus. Early gestation is characterized by differentiation of tissues and organogenesis. From mid-gestation onwards, the focus shifts to accumulation of nutrients, further tissue growth, and organ maturation, preparing the fetus for independent function and postnatal survival. In parallel, early pregnancy is a time of maternal nutrient storage, whereas following mid-gestation, provision of nutrients to the fetus dominates.
MATERNAL METABOLISM
The developing fetus must signal a need for adequate nutrient provision from the mother, in step with its growth requirements. This requires an alteration in maternal food intake and energy handling. In early pregnancy, insulin secretion is increased, with minimal alteration in insulin sensitivity. Insulin resistance is a characteristic feature of late pregnancy and changes the maternal drive from fuel storage to fuel availability. In normal pregnancy, a 50% reduction in insulin-mediated glucose disposal is observed in late gestation.40,41 The genesis of insulin resistance has been attributed to several placentally derived factors.
Despite elevations in both early and late pregnancy, Kirwan et al.35 found no correlation between plasma estradiol, progesterone, prolactin, or hPL and insulin sensitivity during gestation. They noted that the strongest predictor of insulin sensitivity in pregnant women was TNF-α, which significantly negatively correlated with insulin sensitivity in early and late pregnancy, even after adjustment for fat mass. TNF-α increases serine phosphorylation of the insulin receptor substrate, impairing downstream signaling from the insulin receptor and also suppressing adiponectin expression in adipocytes.42 Adiponectin levels decline throughout pregnancy, even in lean women, and circulating levels correlate with the decline in whole body insulin sensitivity.43
Apart from TNF-α, lesser predictors of insulin sensitivity include leptin and cortisol.35 Placental growth hormone is the other major candidate in the genesis of insulin resistance. In mice, overexpression of GHv to levels comparable with that of the third trimester induces significant peripheral insulin resistance.44 GHv increases the expression of the p85α subunit of PI3 kinase in skeletal muscle. This acts as a dominant-negative competitor, preventing p85-p110 heterodimers from binding to IRS-1, effectively producing a postreceptor defect in insulin signaling.45 Elevated levels of p85α protein have also been demonstrated in human studies of pregnancy.42
Insulin resistance promotes substrate availability, but this should not be at excessive maternal expense. To prevent hyperglycemia, detrimental to both mother and fetus, a compensatory increase in insulin secretion occurs.41 Autopsy studies from pregnant women in the latter half of gestation demonstrate enlargement of islet volume and β-cell hyperplasia.46 This process begins early in gestation under the influence of lactogenic hormones.
In animal models, the lactogens increase glucose-stimulated insulin secretion and increase islet cell number and mass. This is achieved through the up-regulation of a number of genes involved in glucose metabolism, insulin gene transcription and synthesis, insulin secretion via exocytosis, cell cycling, and regulation of apoptosis.18,47
Fetal and placental growth in the second and third trimesters generates additional demands for maternal nutrient intake and may provide signals yet to be identified that regulate maternal appetite in proportion to growth requirements. There is some evidence that mothers of male fetuses have higher energy intakes during the second trimester, consistent with the greater growth requirements of male versus female fetuses.48 Prolactin receptors are present in areas of the brain known to be involved in energy balance—the arcuate nucleus, ventromedial hypothalamus, and paraventricular hypothalamic nucleus—and an orexigenic effect of prolactin is apparent in some but not all species.49–51
Maternal subcutaneous fat mass, particularly central, increases during healthy pregnancy.52 Under the influence of estrogen, progesterone, and insulin, lipid accrual in early pregnancy provides a calorie store for mother and fetus. Later in gestation, there is a switch to lipolysis, fat mobilization, and fat oxidation, resulting in increased circulating free fatty acids and a tendency to ketogenesis. These supply maternal energy requirements, sparing glucose and amino acids for the fetus, while also providing steroid hormone precursors to the placenta.53 Triglycerides rise two- to fourfold and cholesterol rises by 25% to 50% during normal pregnancy. The increase in free fatty acids partly reflects resistance to the usual suppression of lipolysis by insulin. Circulating fatty acids in turn contribute to impaired insulin action in peripheral tissues.
Other pregnancy hormones, including prolactin, placental lactogen, growth hormone, and IGFs have effects on adipogenesis and lipolysis in vitro and in rodent/guinea pig models.17,54,55 Prolactin receptors are present in adipocytes and have putative influences on lipoprotein lipase, fatty acid synthesis, and adipokine secretion via signal transducers and activators of transcription (STAT)-mediated pathways.56 The exact contribution of these hormones to lipid metabolism during human pregnancy is still unclear.
FETAL AND PLACENTAL GROWTH AND DEVELOPMENT
Fetal growth results from interactions between the genetic potential of the fetus, maternal nutrition and environment, uteroplacental blood flow/function, and uterine capacity. Hormonal signals convey information between mother and fetus, with the placenta being the main site of communication (Fig. 144-3).
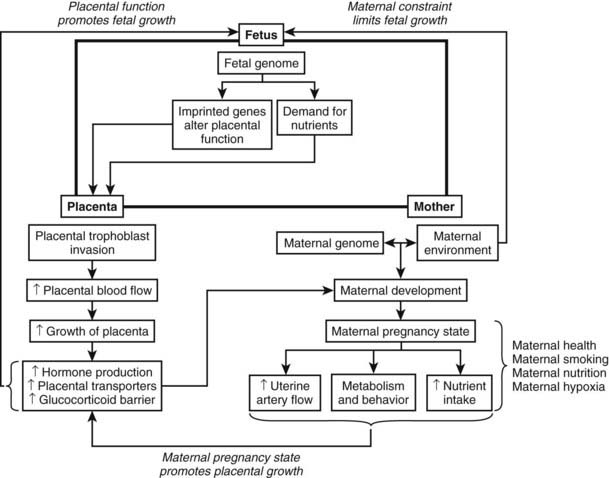
FIGURE 144-3. Interaction between the fetus, placenta, and mother during human pregnancy. Maternal constraint limits fetal growth, whereas the maternal pregnancy state promotes placental growth, which in turn promotes fetal growth. The fetus and mother communicate via the placenta.
(Data from Murphy VE, Smith R, Giles WB, et al: Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus, Endocr Rev 27:141–169, 2006.)
While growth hormone is a critical regulator of postnatal growth, it has little role in the fetal period, when IGFs directly control growth in a tissue-specific and time-specific manner. IGF-1 and IGF-2 are expressed in fetal tissues from very early in gestation, and their receptors have been located in numerous cell types. IGFBPs are also expressed widely in fetal tissues and locally modulate the effects of the IGFs.34
In humans, the IGF2 gene is more abundantly expressed in fetal tissues than IGF1, and fetal plasma concentrations of IGF-2 are 3 to 10 times higher than IGF-1 during late gestation. After delivery, levels of IGF-1 rise under the influence of GH, and IGF-2 declines, indicating a shift in the mechanisms of growth regulation at the time of transition from placentally derived to enterally derived nutrition.57 In mice, IGF-1 directly affects fetal growth, whereas IGF-2 also impacts on placental development and nutrient transfer. Deletion of either Igf-1 or Igf-2 resulted in similar decreases in birthweight of about 40%.58 A combined deletion of both Igfs resulted in an additive reduction of 70%.59 Mice with Igf-2 deletion additionally have smaller and structurally abnormal placentae, while Igf-2 overexpression results in fetal overgrowth and placentomegaly.60,61 Manipulations in the Igf-1 and Igf-2 genes result in abnormal development of individual tissues as well as overall changes in body weight and size, consistent with the hypothesis that these growth factors have tissue-specific roles in cell growth, differentiation, and apoptosis.
In humans, a homozygous partial deletion of the IGF1 gene resulted in both intrauterine and postnatal growth retardation, accompanied by sensorineural deafness and mental retardation.62 Abnormalities in the IGF-1 receptor gene are also linked to intrauterine growth retardation.63 Birthweight is positively associated with cord blood IGF-1 and inversely related to IGFBP-1 in normal term infants (reviewed in Ref. 34). A positive association between IGF-2 and placental weight has also been reported.64
Animal studies lend further insight into the mechanisms by which environmental and nutritional factors interact with the IGF system. Experimentally induced reductions in maternal nutrient availability, oxygen supply, uterine blood flow, or placental function predominantly decrease fetal IGF-1 concentrations, with minimal or no effect on IGF-2.57 In fetal sheep, infusions of glucose increase IGF-1 levels, demonstrating a relationship between nutrient availability and the growth response. In sheep, IGF-1 levels positively correlate with insulin concentrations and are reduced by thyroid hormone deficiency. In contrast, glucocorticoids down-regulate Igf-2 gene expression, suggesting a potential mechanism by which conditions of intrauterine stress could restrict growth.65 The surge in cortisol levels in the weeks leading up to parturition is proposed to result in a switch from fetal IGF-2 to IGF-1 predominance in neonatal life. Expression of mRNA and plasma levels of IGFBP-1 also respond to experimentally induced alterations in fetal nutrient supply (reviewed in Ref. 57).
Human studies demonstrate alterations in the IGF system with conditions of in utero deprivation.66 Reductions in IGF-1, IGF-2, and elevations in IGFBP-1 have been variably described in cases of intrauterine growth retardation.34 It remains unclear how the maternal environment alters IGF signaling, but the important contribution of the IGF axis to fetal growth is evident.
Fetal insulin is secreted by the pancreas from 11 to 15 weeks of development and is an important link between nutrient availability and fetal growth. Insulin has an anabolic effect on fetal tissues both directly and through stimulation of IGF-1. Unlike IGF-1, it promotes tissue accrual without effects on cell differentiation. Fetal insulin secretion is responsive to glucose, as sensed by the glucokinase enzyme, and mutations in its encoding gene raise the set-point at which glucose stimulates insulin secretion, resulting in mild blood glucose elevation.67 In mouse models of this mutation, fetuses born to normoglycemic mothers have lower birthweights, consistent with reduced insulin production; however, those born to hyperglycemic mothers have normal birthweight.68 These data indicate that maternal nutrient availability results in signals that increase fetal insulin production and hence tissue growth. Thus, fetal hyperinsulinemia places infants from hyperglycemic pregnancies at risk of macrosomia.
Placentally derived factors promote placental development in an autocrine/paracrine fashion. Both GHv and IGF-2 have been linked to invasiveness of the extravillous cytotrophoblast.69,70 In vitro, IGF-1 and IGF-2 enhance human cytotrophoblast proliferation and inhibit apoptosis, acting via the IGF-1 receptor and MAP kinase and PI3 signaling pathways.71 Enhancement of IGF-1 and IGF-2 levels in human primary placental fibroblasts by adenoviral transfection improved proliferation, migration, and survival, while reducing levels had an opposite effect.72 IGF-2 further promotes placental angiogenesis and uterine vascular remodeling.73
The IGF2 gene is paternally imprinted in the placental and other fetal tissues. It is one of a number of imprinted genes involved in fetal and placental development. According to the genetic conflict theory, genes that are paternally expressed favor nutrient extraction and growth of the offspring, conferring greater likelihood of postnatal survival and reproduction. Maternally expressed genes curb excessive growth in order to preserve maternal health, facilitate parturition, and maximize ongoing reproductive potential.74 The placental expression of Igf-2 is consistent with this theory. Heterozygous deletion of a transcript of this gene expressed specifically in the murine labyrinthine trophoblast resulted in placentae that were smaller with reduced passive diffusion capacity per gram and a compensatory increase in active placental amino acid transport, so that fetal growth was initially maintained but fell off late in gestation.75 In contrast, the Igf-2 receptor and h19 genes are maternally imprinted and serve as control mechanisms for IGF-2. The IGF-2 receptor binds IGF-2 and limits its activity, while maternal H19 prevents the bi-allelic expression of Igf-2. Loss of either of these genes results in fetal overgrowth.76 Hence, imprinting of these and multiple other genes in the placenta regulates fetal growth.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


