Chapter Outline
HUMAN HEMOGLOBINS: COMPOSITION AND GENETICS
CLASSIFICATION OF THE THALASSEMIAS
Origin of Thalassemia Mutations: The Influence of Malaria
Classes of Mutations That Cause Thalassemia
Identification, Characterization, and Ethnic Distribution of β-Thalassemia Mutations
Mutations That Affect β-Globin Gene Regulation
β-Thalassemia Mutants Unlinked to the β-Globin Complex
Hereditary Persistence of Fetal Hemoglobin
Mutations That Alter α-Globin Gene Regulation
Deletion Mutations within the α-Globin Gene Cluster
Mutations Not Linked to the Globin Gene Clusters That Alter Globin Gene Expression
CLINICAL HETEROGENEITY OF THALASSEMIA SECONDARY TO DIVERSITY OF MUTATIONS
The thalassemias are a heterogeneous group of inherited forms of anemia caused by mutations that affect the synthesis of hemoglobin. Milder forms are among the most common genetic disorders, whereas the severe forms, which are seen less often, lead to significant morbidity and mortality worldwide ( Fig. 21-1 ).
Study of the thalassemias traces the history of the application of recombinant deoxyribonucleic acid (DNA) methods to analysis of inherited diseases and underscores how naturally occurring mutations in humans illuminate genetic principles. In this chapter the genetics of hemoglobin genes are reviewed as background for discussion of the molecular basis of the thalassemia syndromes, their clinical phenotypes, current management, and prenatal diagnosis.
Human Hemoglobins: Composition and Genetics
Normal hemoglobins are tetramers of two α-like and two β-like globin polypeptides. The predominant hemoglobin in normal adult red blood cells is hemoglobin A (HbA), α 2 β 2 . The α- and β-globins contain 141 and 146 amino acids, respectively. In addition to HbA, adult red blood cells normally contain two minor hemoglobins: HbA 2 (α 2 δ 2 ) and fetal hemoglobin (HbF) (α 2 γ 2 ). The γ and δ polypeptides are related to β but differ in their primary amino acid sequences; hence they are referred to as β-like globins. HbA 2 normally accounts for 2% to 3.5% of the total hemoglobin. Although it is a minor component in adult red blood cells, HbF is the predominant hemoglobin in fetal red blood cells during the latter two trimesters of gestation. Because it does not bind 2,3-diphosphoglycerate, its affinity for oxygen is higher than that of HbA. As such, HbF enhances the fetus’ ability to extract oxygen from the placenta. HbF constitutes a small fraction of the total hemoglobin in adult red blood cells (0.3% to 1.2%), in which it is largely restricted to a small subset of circulating erythrocytes (0.2% to 7% of the total cells), termed F cells . Production of HbF in normal adults is largely genetically controlled as demonstrated by studies in twins and familes. During the first trimester in utero, embryonic hemoglobins with differing subunit composition are found in the yolk sac–derived macrocytic (or primitive) red blood cells.
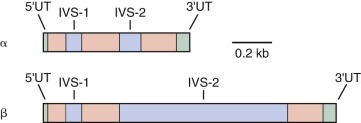
The genes that encode the globin polypeptides are organized into two small clusters. The α-like genes are located near the telomere of the short arm of chromosome 16 (16p13.3), whereas the β-like genes reside on chromosome 11 at band 11p15.5. A schematic diagram of the human globin genes and the composition of the various hemoglobins are shown in Figure 21-2 .
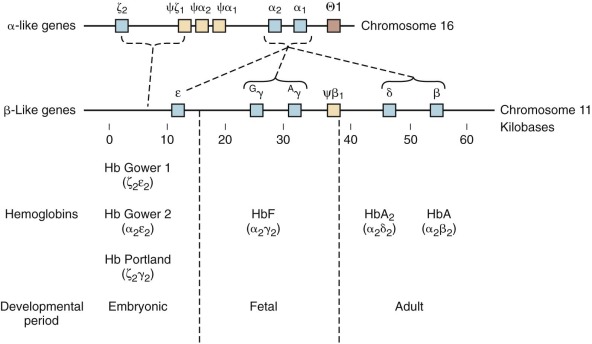
The α-globin gene cluster contains three functional genes, ζ, α 2 , and α 1 , oriented in a 5′-to-3′ direction along the chromosome. ζ-Globin, encoded by the ζ gene, is found in two embryonic hemoglobins, Hb Gower-1 (ζ 2 ε 2 ) and Portland (ζ 2 γ 2 ). The duplicated α-globin genes (α 1 and α 2 ) encode identical polypeptides. DNA sequence analysis has revealed three additional globin gene–like sequences in the cluster: pseudo-ζ (ζ 1 ), pseudo-α 1 , and pseudo-α 2 . Although they resemble the functional genes, sequence differences in coding or critical regulatory regions render these genes inactive, and hence they are referred to as pseudogenes.
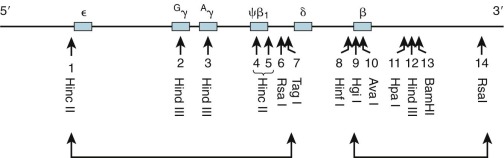
Five functional genes, ε, G γ, A γ, δ, and β, are present within the β-like cluster and arranged in a 5′-to-3′ direction as they are expressed during development. The product of the embryonic ε gene is found in the embryonic hemoglobins Hb Gower-1 (ζ 2 ε 2 ) and Hb Gower-2 (α 2 ε 2 ). The fetal γ genes are duplicated but encode globins differing only at amino acid 136; G γ-globin and A γ-globin contain a glycine or alanine residue, respectively. G γ- and A γ-globins are both found normally in HbF (α 2 γ 2 ). The δ-globin gene encodes a polypeptide differing in only 10 of 146 residues from β, and yet it is expressed at a very low level in adult red blood cells (<3% of β). The poor expression of δ-globin is attributed to differences in critical regulatory sequences within the gene that appear to inhibit messenger ribonucleic acid (mRNA) processing and the inherent instability of δ mRNA. Only a single functional β-globin gene is present in the cluster. β-Globin is the predominant β-like globin in adult red blood cells, in which HbA (α 2 β 2 ) accounts for more than 95% of the total hemoglobin.
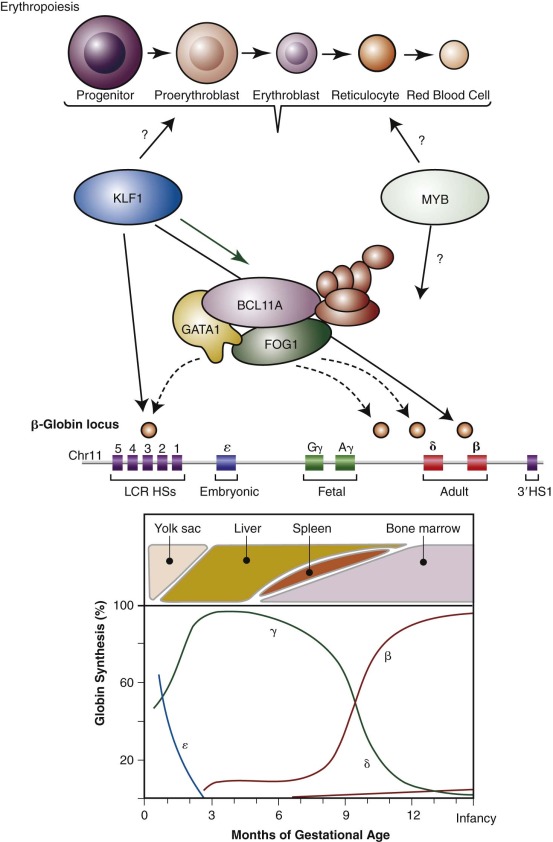
The relative synthesis of individual globin chains, the major sites of erythropoiesis during development, and major regulators of this process (discussed later) are depicted in Figure 21-3 . Embryonic hemoglobins are expressed nearly exclusively in primitive nucleated red blood cells differentiating in the yolk sac blood islands. HbF production commences during the next wave of erythropoiesis in the fetal liver. Fetal liver–derived red blood cells lose their nuclei as terminal maturation occurs, whereas the primitive, yolk sac–derived cells remain nucleated. The transition from HbF to HbA coincides approximately with the switch from fetal liver to bone marrow erythropoiesis. Despite this correlation between the site of erythropoiesis and the hemoglobins expressed, careful analysis of tissues derived from experimental animals and human fetuses has shown that embryonic hemoglobins are synthesized in the liver as well as the yolk sac and that HbF is produced in the bone marrow as well as the liver. The developmental switches in hemoglobin expression are related to the time of gestation rather than the anatomic site of erythropoiesis per se.
Globin Gene Structure
The globin genes were among the first eukaryotic genes isolated by recombinant DNA cloning methods in the late 1970s. Subsequent work provided the entire DNA sequences of the human α- and β-globin gene clusters and extensive sequences of other vertebrate globin complexes. These data have been invaluable in determining the mutations underlying thalassemia syndromes and in manipulating gene regulatory regions.
Intervening Sequences or Introns
A remarkable finding was made on initial study of the globin genes: the coding region, rather than being organized in a single continuous unit, is interrupted by noncoding DNA known as intervening sequences (IVSs), or introns. Most eukaryotic genes contain one or more introns. As indicated in Figure 21-4 , globin genes are interrupted at two positions. The discontinuous nature of the coding region of globin genes poses a formidable problem for the formation of mRNA that must be translated into globin polypeptides on cytoplasmic ribosomes. Transcription of a globin gene generates a precursor mRNA containing introns. Formation of mature mRNA is accomplished by posttranscriptional processing, termed RNA splicing . The pathway of RNA processing is depicted in Figure 21-5 .
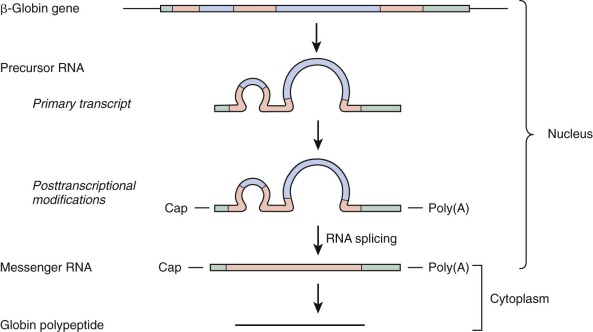
RNA splicing must be executed with exquisite precision for functional mRNA to be generated. Because translation of mRNA proceeds by the reading of triplets (codons), excision of introns needs to be accurate to the nucleotide; otherwise, shifts in the reading frame of the translated polypeptide will result. RNA processing is guided by specific sequences, known as splice site consensus sequences, located at the 5′ and 3′ boundaries of the introns. The donor site, which marks the 5′ end of the intron, generally conforms to the sequence 5′ (C/A)AG′ GT (A/G)AGT, where the prime sign indicates the position of splicing and GT is an essentially invariant dinucleotide at the 5′ end of the intron. The acceptor site, which defines the 3′ end of the intron, usually fits the consensus 5′ (T/C) n N(C/T) AG ′G, where n is 11 or greater, N is any nucleotide, the prime sign indicates the site of splicing, and AG is an essentially invariant dinucleotide. Excision of introns generally occurs between the dinucleotides GT and AG, known as the GT-AG rule.
Conserved Features of Mature Globin mRNA
Sequences of human globin genes represented in processed globin mRNA include additional segments located before (5′) and after (3′) the coding region. These untranslated regions are depicted in Figure 21-5 . In addition, the mature mRNA is modified at both termini. At the 5′ end, a methylated guanylic acid (m 7 G) cap structure is present. A variable number of adenylic acid residues are added at the 3′ end and constitute a poly(A) tail. The 5′ cap structure appears to be important for efficient initiation of mRNA translation, whereas the poly(A) tail contributes to mRNA stability. Overlapping the beginning of the mRNA sequence in genomic DNA are sequences that aid in directing the initiation of transcription to the proper site. In some eukaryotic genes these sequences conform to an initiator (Inr) consensus element. The human β-globin gene has been shown to possess an Inr element that is functional in transcription reactions performed in vitro.
Polyadenylation at the 3′ end of mRNA precursors depends on a signal in the 3′-untranslated region, generally AAUAAA (AATAAA in genomic DNA). The mechanism of modification of the 3′ end is complex and involves not only polyadenylation but also cleavage of the precursor RNA because the primary RNA transcript extends several hundred nucleotides past what becomes the position at which the poly(A) tail is added.
Translation of mRNA into a polypeptide proceeds by the reading of triplets (codons) on cytoplasmic ribosomes. The first AUG codon (specifying methionine) present in the mRNA specifies the start site for translation of the mRNA into protein and is embedded in a sequence context (the Kozak consensus sequence, typically CC[A/G]CC ATG G) that signals the binding of translation initiation factors and ribosomes to the mRNA. Usually the amino-terminal methionine residue is removed from the growing polypeptide chain even before its synthesis is completed. Termination of polypeptide chain translation is directed by the termination codons UAA, UAG, or UGA. Mutation of these codons allows continued translation into the 3′-untranslated sequences of mRNA, as occurs in selected α-globin chain variants associated with α-thalassemia (discussed later).
As briefly reviewed earlier, the formation of functional mature mRNA demands extraordinary precision and depends on highly conserved sequence elements. As exemplified by the thalassemia syndromes, point (or other) mutations in these signals lead to reduced or absent polypeptide chain synthesis, which is the hallmark of thalassemia. Mutations causing thalassemia involve all phases of gene expression, including gene transcription, RNA splicing, integrity of the coding sequence, 3′ polyadenylation, and translation initiation (discussed later).
Regulation of Globin Gene Expression
Globin genes in all vertebrates are expressed in a tissue-specific and developmentally programmed manner. Their transcription is activated only within developing erythroid precursor cells. Moreover, individual globin genes are expressed at different developmental stages. Hence within the genes of the β cluster, globins are expressed at the embryonic (ε), fetal (γ), or adult (β and δ) stages, whereas within the α cluster, embryonic (ζ) and adult (α) chain expression is seen. A central problem posed by the organization of the globin gene clusters is how these patterns of tissue- and developmental-stage specificity are achieved. Recent findings from human genetic studies have begun to elucidate the molecular control of this developmental switch in the β-globin gene cluster and are discussed later in this chapter.
Proximal Regulatory Sequences and Transcription Factors
Several conserved sequence elements (motifs) in the 5′-flanking sequences of globin genes comprise the promoter, a region required for accurate and efficient transcription of genes by RNA polymerase II. Promoters of vertebrate globin genes are similar in overall configuration and subset of motifs present but differ in their detailed organization and sequences. Promoters generally cooperate with more distant regulatory elements, termed enhancers, to stimulate transcription. As discussed later, globin gene promoters appear to interact in a synergistic fashion with very powerful distant elements known as locus control regions (LCRs).
The TATA (or ATA) box is a motif seen in nearly all promoters, including those of the globin genes. The TATA box, typically located 20 to 30 base pairs (bp) upstream from the transcription start site, constitutes the binding site for a general transcription factor, the TATA-binding protein (TBP). Binding of TBP to the TATA box is the first step in the assembly of a basal transcription complex (often termed TFIID ) that includes many additional proteins (such as TFIIA, TFIIB, TFIIE/F, and TFIIH) and RNA polymerase II. Mutations within the TATA box, as occur in some types of β-thalassemia, decrease the binding of TBP to the promoter and decrease transcription.
DNA sequence motifs located upstream of the TATA box bind proteins that interact with the general transcription machinery through protein-protein contacts with the TFIID complex and other associated proteins. These promoter-bound proteins may either increase (activate) or decrease (repress) the rate of transcription. A small set of motifs are consistently present in globin gene promoters, including the CCAAT box, the CACC box, and GATA consensus sequences. Each motif may be viewed as a potential binding site for one or multiple transcription factors, which are either tissue restricted or ubiquitous in their cellular distribution.
Transcription factors are typically viewed as modular proteins made up of two domains that fulfill different functions: a DNA-binding domain responsible for sequence-specific DNA recognition and an activation (or repression) domain or domains that interact with components of the basal complex to modulate transcription. Additionally, some transcription factors may function to recruit activities that modify the histones around which DNA is wrapped to make chromatin. These modifications can indirectly alter the activity of the basal transcriptional complex. It is currently believed that transcriptional specificity is achieved by functional cooperation and interaction between cell-restricted and general transcription factors. As background for understanding globin gene control, the presently characterized erythroid-enriched transcription factors are reviewed here. For additional discussion of these proteins, readers are referred elsewhere.
The consensus motif (A/T)GATA(A/G), known as the GATA motif, is found in the promoter region of most vertebrate globin genes and binds an abundant erythroid-restricted transcription factor, GATA1. GATA motifs have been identified in the regulatory elements of virtually all erythroid-expressed genes, consistent with the notion that GATA1 should serve a critical role in erythroid gene expression. As noted later, multiple GATA sites are also present within distant regulatory elements. The essential role of GATA1 in erythroid development was formally demonstrated through gene targeting experiments in mouse embryonic stem cells. Disruption of the single X-chromosome GATA1 gene in totipotent embryonic stem cells prevents their development into normal erythroid cells. Naturally occurring but rare hypomorphic mutations of GATA1 in humans have been shown to cause β-thalassemia or disrupt normal erythroid development. The GATA1 protein is a member of a small family of related “GATA factors” that are distinguished by a novel zinc-finger DNA-binding domain. In addition to merely specifying DNA recognition, this domain also mediates protein-protein interactions. Accordingly, GATA1 is able to interact physically with other GATA1 molecules or with other types of zinc-finger proteins, including the ubiquitous CACC- or GC-binding factor Sp1, the transcription factor erythroid Krüppel-like factor (EKLF), and a specific cofactor called FOG-1 (for “friend of GATA1” ; discussed later). It is envisioned that through its multiple physical interactions, GATA1 cooperates with other transcription factors, perhaps bound to DNA at distant sites, to program erythroid-specific transcription.
CACC motifs, which are represented by diverse sequences within globin and other gene promoters, bind a variety of transcription factors. Many CACC sequences are recognized by Sp1, a ubiquitous zinc-finger activator protein. A particular CACC motif, CCACACCCT, is found in the adult β-globin gene promoter and is recognized with high affinity by the erythroid-specific protein EKLF (also known as KLF1). The functional relevance of this binding site has been established through naturally occurring mutations that lead to β-thalassemia (discussed later). In addition, gene targeting (or knockout) experiments in mice have formally established that EKLF/KLF1 is necessary for efficient β-globin transcription in vivo.
A third erythroid transcription factor, known as NF-E2, binds to an extended motif—(T/C)TGCTGA(C/G)TCA(T/C)—that is found within some distant regulatory elements (discussed later) and a small subset of erythroid promoters but not within globin gene promoters. NF-E2 is a heterodimer of two polypeptides of the basic domain–leucine zipper (or “b-zip”) class of transcription factors. One subunit of NF-E2 is tissue restricted, whereas the other is ubiquitous. Although NF-E2 is essential for globin gene expression in mouse erythroleukemia cells in tissue culture, its role in vivo appears to overlap that of one or more unknown factors that may act through the same target sites in DNA.
Locus Control Regions and Chromatin Domains
How is globin gene transcription activated and developmentally controlled? Inspection of the DNA sequences of globin gene promoters in the early 1980s failed to provide substantive insight. Initial attempts to dissect control elements involved introducing globin genes into the germline of mice by oocyte injection but were plagued by low-level and erratic transgene expression. Nonetheless, it was possible to show that low-level stage specificity was imparted by the human β- and γ-globin gene promoters. For example, when the human β-globin promoter is introduced into transgenic mice, it directs gene expression only in adult erythroid cells, whereas the human γ-globin promoter is active only in embryonic erythroid cells (mice do not have an HbF stage).
These early globin gene regulation studies suggested that critical regulatory elements required for high-level expression were missing from the immediate vicinity of the genes themselves. When sensitivity to digestion by the enzyme DNase I was used as an indicator of chromatin structure in the mid 1980s, regions of extreme sensitivity (hypersensitivity sites [HSs]) were identified far upstream (≈30 to 50 kilobases [kb]) of the adult human β-globin gene. Four subregions were delimited; they are present in the chromatin of erythroid, but not in nonerythroid cells. An additional site located even further upstream was found in all tissues. In a formal test of their functional relevance, the HSs were linked to a human β-globin and introduced into the germline of mice. Remarkably, transgenic mice then expressed the human β-globin gene at a level equivalent to that of the endogenous mouse β gene. Further studies showed that the transgene is expressed not only in a tissue-specific manner but also in a copy number–dependent fashion, independent of the chromosomal site of integration. These HSs comprise an essential distal regulatory domain, now referred to as the LCR (see elsewhere for a more detailed history and discussion).
Other globin gene clusters also contain erythroid-specific DNase I HSs. A segment of extreme DNase I hypersensitivity (known as HS-40) located far upstream of the human α-globin genes serves as an enhancer for the α locus. HS-40, however, does not display the full properties of an LCR because it does not direct copy number–dependent transgene expression. Nonetheless, the in vivo relevance of both the β-LCR and HS-40 is underscored by the discovery of patients with thalassemia who have specific deletions in these regions (discussed later).
The human β-LCR, HS-40, and analogous regions studied in other species are composed of cores, each encompassing a DNase I HS. Cores are approximately 200 to 300 bp in length. Remarkably, within the cores three major protein-binding sites are consistently found: GATA, AP-1 (NF-E2), and CACC sequences. The position-independent activity of the β-LCR correlates best with the presence of GATA and CACC motifs, particularly within the subregion known as HS-3. Enhancer activity of the LCR, particularly within subregion HS-2, requires the NF-E2 motif. The protein-binding motifs within the LCR are also found in globin and other erythroid-expressed gene promoters. To date, no protein-binding sites unique to LCR elements have been identified. Hence the distinctive properties of the LCR (or HS-40) appear to reflect the synergistic interactions of more typical transcription factors rather than the action of a new set of regulatory proteins.
The discovery of distant control elements, marked by DNase I hypersensitivity, emphasizes the relationship between chromatin structure and globin gene regulation, an association solidified by the unraveling of a rare syndrome, α-thalassemia with X-linked mental retardation. This condition results from mutations in a gene designated XH2 (or ATRX ) that encodes a member of the helicase superfamily. Such proteins, which are often involved in DNA recombination and repair and in the regulation of transcription in Drosophila , yeast, and mammals, appear to influence transcription in a global manner by altering chromatin structure.
Regulation at a Distance: Globin Gene Switching
How do LCR sequences influence globin gene transcription over large distances (>50 kb), and how are the individual globin genes developmentally regulated? Two formal possibilities have been considered. On one hand, the LCR might merely provide an environment conducive for activation of the downstream globin genes. The globin genes would be autonomously regulated; that is, the developmental profile of their expression would be intrinsic to the individual genes (and presumably determined by their promoters). The “influence” of the LCR is most simply viewed as reflecting physical association of the LCR with globin genes brought into apposition by chromosomal looping. On the other hand, sequential activation of the particular genes might depend (at least in part) on competition of each gene for influence of the LCR such that only one gene-LCR interaction would be productive on a single chromosome at any time. The outcome of the competition would be dependent on the array of proteins bound not only at each promoter but also at specific sites in the LCR. Data in favor of both autonomous and competitive mechanisms of regulation have been obtained (see Orkin for a detailed discussion).
The human embryonic ζ- and ε-globin genes appear to be largely autonomously regulated. Transgenes that contain LCR are expressed during embryonic erythropoiesis (the yolk sac stage) and then extinguished during the fetal liver stage. The information required for shutoff is contained near the globin genes, and competition by adjacent globin genes is not required. Shutoff is hypothesized to reflect the action of repressors, or silencer proteins, that bind the gene promoters. Motifs within the human ε-globin promoter are involved in silencing bind GATA1 and a ubiquitous factor, YY1.
The competitive model of gene regulation is based on experiments in chicken red blood cells that demonstrate competition between the chicken β- and ε-globin gene promoters for a single enhancer located between the genes. In the chicken it has been proposed that an adult stage–specific factor (NF-E4) favors interaction of the β promoter with the enhancer to the exclusion of the ε promoter. In an analogous fashion, data suggest that the human β-globin gene may be negatively regulated in a competitive fashion by the γ-globin gene. Whereas the γ gene is largely autonomously regulated, the β-globin gene is silenced in the embryonic and early fetal stage by a linked γ gene (a γ gene in cis to β). Shutoff of the γ gene, presumably as a result of repressors (or silencers), allows the adult β-globin to be expressed. Although other models are theoretically possible, the capacity of the LCR to act at a distance in regulating activation of globin genes is most compatible with the formation of physical contacts between the LCR (or subregions thereof) and their associated proteins with regulatory elements neighboring the genes themselves. Stage-specific and competitive regulation would therefore reflect engagement of the LCR with genes one at a time. LCR-gene interactions probably have intrinsic stabilities and off-rates such that a single erythroid cell might express more than one globin over time, even from a single chromosome. Experiments examining nascent human globin RNA along the β-gene complex tend to support such models and lend credence to the notion that chromosomal looping brings the LCR and individual genes in apposition. Evidence for LCR interactions with the proximal promoter regions of both the β- and α-globin genes has recently been directly demonstrated by using novel in vivo biochemical approaches. Dynamic interactions between the β-LCR and the γ- and β-globin genes appear to underlie the reciprocal expression of these genes in erythroid cells and provide hope that subtle alterations in the nuclear environment may facilitate reactivation of γ-globin genes in patients with hemoglobinopathies such as sickle cell anemia or β-thalassemia. Recent work suggests that looping of the LCR itself in close proximity to the globin genes may be sufficient to allow for at least partial activation of transcription of these genes, as least in mouse models. This group has recently gone on to demonstrate that such modulation of looping is sufficient for activation of embryonic or fetal hemoglobin genes in mouse models and human cells. This offers the prospect that such approaches could be therapeutically useful.
Recent Insights into Developmental Globin Gene Switching from Human Genetics
The molecular basis of the fetal-to-adult hemoglobin switch had remained an enigma over the course of nearly three decades. Although regulators of globin gene regulation, including GATA1, EKLF/KLF1, and NF-E2, had been identified, none of these regulators appeared to be sufficient to confer the stage-specific expression properties that are observed for the γ- and β-globin genes. By following up on the results of genome-wide association studies (GWAS)—a technique by which associations between common polymorphisms across the genome and various traits are tested—the first developmental stage-specific regulator of the fetal-to-adult hemoglobin switch, BCL11A, was identified (see Fig. 21-3 ). Initial GWAS had identified a robust signal of association on chromosome 2 with HbF levels in both healthy persons and in patients with sickle cell disease. This signal was most robust within a regulatory element of the gene BCL11A , which encodes a multizinc finger transcriptional regulator that had previously been implicated in B lymphocyte and neural development. By examining the hypothesis that BCL11A may also regulate HbF expression, it was found that reducing BCL11A expression allowed robust reactivation of HbF in adult cells. Consistent with this finding, BCL11A is developmentally regulated and its expression is maximal in erythroid cells with little or no γ-globin expression. Moreover, BCL11A occupies regulatory elements within the β-globin locus, although it does not bind the promoter of the γ-globin genes. BCL11A holds tremendous promise as a therapeutic target for HbF induction in patients with β-thalassemia or sickle cell disease. Indeed, a proof of concept study has shown that removal of BCL11A in the hematopoietic system of mouse models of sickle cell disease can result in amelioration of disease in this model system. An erythroid-specific enhancer element of BCL11A has recently been identified and holds promise for tissue-specific targeting of BCL11A expression. Furthermore, recent insight from rare human deletions within the β-globin gene cluster, which will be discussed in more detail later in this chapter, are providing important mechanistic insight into how BCL11A acts to regulate the fetal-to-adult hemoglobin switch. It is likely that further studies of the type described here will lead to targeted approaches by which BCL11A activity can be selectively modulated.
In addition to the insight gained into hemoglobin switching from studies of BCL11A, other work has suggested that rare mutations in KLF1/EKLF may lead to elevations of HbF levels in patients and that this transcription factor may be involved in hemoglobin switching through the regulation of BCL11A (see Fig. 21-3 ). However, other mutations in KLF1 result in either a significant congenital anemia or have little affect upon globin gene regulation, suggesting that other factors may modify the observed phenotypes in the patients. Therefore further study of KLF1 and its role in this regulatory process is needed. In addition to the GWAS highlighting the role of BCL11A in HbF regulation, these studies have also suggested a role for the MYB gene in silencing of HbF. This role has been substantiated through functional studies that have shown that reducing MYB levels can robustly activate γ-globin expression. GWAS of β-thalassemia patient populations have shown that variants upstream of MYB can have a profound affect on the clinical course, and therefore MYB may be an important therapeutic target. However, further studies are needed to better understand the exact mechanism by which MYB regulates globin gene expression (see Fig. 21-3 ).
Classification of the Thalassemias
The hallmark of thalassemia syndromes is decreased (or absent) synthesis of one or more globin chains. The designation α- and β-thalassemia refers to deficits in α- and β-globin production, respectively. The α- and β-thalassemias include clinical syndromes of varying severity ( Table 21-1 ). Knowledge of molecular genetics provides a framework in which to consider their clinical heterogeneity.
| Clinical Classification | Description |
|---|---|
| Silent carrier (α or β) | Hematologically normal |
| Thalassemia trait (α or β) | Mild anemia with microcytosis and hypochromia |
| HbH disease (α-thal) | Moderately severe hemolytic anemia, icterus, and splenomegaly |
| Hydrops fetalis (α-thal) | Death in utero caused by severe anemia |
| Severe β-thalassemia (Cooley anemia) | Severe anemia, growth retardation, hepatosplenomegaly, bone marrow expansion, and bone deformities |
| Thalassemia major | Transfusion dependent |
| Thalassemia intermedia | No regular transfusion requirement |
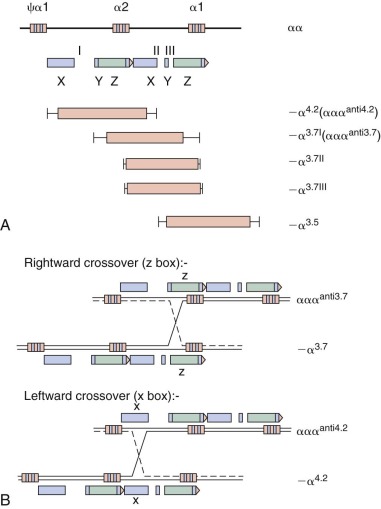
Because the structural gene for α-globin is duplicated on chromosome 16, each diploid cell contains four copies of the α-globin gene. The four α-thalassemia syndromes—silent carrier, α-thalassemia trait, hemoglobin H (HbH) disease, and hydrops fetalis (see Table 21-1 )—reflect the inheritance of molecular defects affecting the output of one, two, three, or four of the α-globin genes, respectively. More than 30 different mutations affecting one or both α-globin genes on a chromosome have been described. Some mutations abolish expression of an α-globin gene (α 0 ), whereas others reduce expression of the gene to a variable degree (α + ). Marked genetic and clinical heterogeneity occurs within the four general categories of α-thalassemia. Heterogeneity arises because the syndrome in any given person may represent a combination (or so-called interaction) of 2 of the 30 or more mutations that have been described.
The β-thalassemias also include four clinical syndromes of increasing severity—silent carrier, thalassemia trait, thalassemia intermedia, and thalassemia major (see Table 21-1 ). In contrast to the α-thalassemias, the four classes of β-thalassemia are not correlated with the number of functioning genes. Because a single functional β-globin gene resides on each chromosome 11, a diploid cell normally has two β-globin genes. The clinical heterogeneity of the β-thalassemias represents the diversity of specific mutations that variably affect β-globin gene expression. Almost exclusively, these mutations involve the β-globin gene rather than an unlinked genetic determinant. Many mutations eliminate β-gene expression (β 0 ), whereas others cause a variable decrease in the level of β-gene expression (β + ). The capacity of individual patients to synthesize γ-globin modulates the clinical severity. Such is the case because the severity of thalassemias is determined by the degree of globin chain imbalance rather than by the absolute level of either α- or ββ-globin synthesis per se. Substantial synthesis of γ-globin in the marrow cells of persons with β-thalassemia lessens the extent of chain imbalance and therefore improves red blood cell production. Particular mutations of the β-globin gene in β-thalassemia appear to affect γ-globin gene expression directly. However, some persons with otherwise severe β-thalassemia may co-inherit additional genetic determinants that enhance the synthesis of HbF. Indeed, in patients with β-thalassemia who do not receive transfusions, HbF levels are strongly associated with clinical severity. Coincident inheritance of an α-thalassemia mutation also reduces chain imbalance in patients with homozygous or heterozygous β-thalassemia. Clinical severity in any individual patient represents the outcome of these complex genetic interactions.
Origin of Thalassemia Mutations: The Influence of Malaria
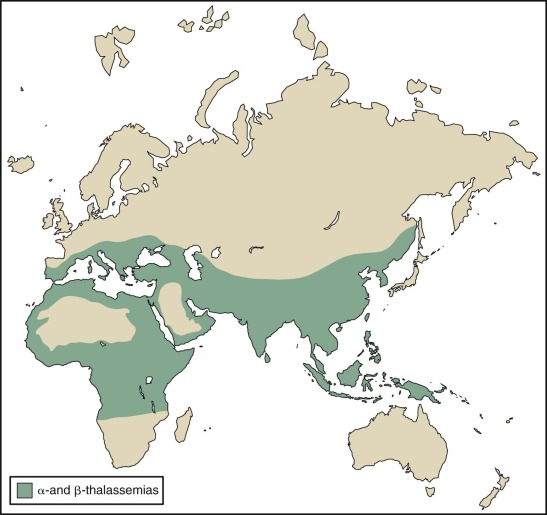
Mutations causing thalassemia have arisen spontaneously. The nearly exclusive distribution of lethal red blood cell disorders such as thalassemia, sickle cell disease, and glucose 6-phosphate deficiency in tropical and subtropical regions led Haldane in 1949 to propose that the heterozygous carrier state for these conditions confers a selective advantage in locations where malaria is endemic. The incidence of these genes in a population is determined by the balance between premature death of homozygotes and increased fitness of heterozygotes. The frequency of β-thalassemia mutations is high (>1%) in regions such as the Mediterranean basin, northern Africa, Southeast Asia, India, and Indonesia but uncommon in northern Europe, Korea, Japan, and northern China. The incidence of β-thalassemia trait may exceed 20% in some villages in Greece. α-Thalassemia is perhaps the most common single gene disorder in the world. The frequency of α + -thalassemia alleles ranges from 5% to 10% in the Mediterranean basin, 20% to 30% in portions of West Africa, and 68% in the southwest Pacific. The incidence of α-thalassemia is less than 1% in Britain, Iceland, and Japan. Although the incidence of malaria and the rate of occurrence of thalassemia are not always inversely correlated, the anomalies and inconsistencies seem to be the result of genetic drift, migration, and demographic changes that have occurred in the last 10,000 years.
Additional epidemiologic studies have provided further evidence for the validity of the “malaria hypothesis” in both α- and β-thalassemia. Siniscalco and colleagues showed that β-thalassemia is uncommon in inhabitants of the mountainous areas of Sardinia, where malaria is rare, compared with the incidence in coastal populations. In Melanesia, α-thalassemia is correlated with malaria across both latitude and altitude. β-Thalassemia in Melanesia is also associated with malarious coastal regions. Williams and colleagues found that children with α-thalassemia trait appear to have a higher incidence of malaria in childhood that appears to confer subsequent immunity to more severe malarial infections. Follow-up studies in other populations have demonstrated that although α-thalassemia does not confer a reduced risk of malarial infection, it does dramatically reduce the incidence of severe malarial complications.
The cellular mechanisms responsible for the selective advantage of thalassemia heterozygotes remain incompletely defined. Cultured erythrocytes containing high concentrations of HbF retard the growth and development of Plasmodium falciparum . β-Thalassemia heterozygotes have a delayed disappearance of HbF in the first year of life, which might provide protection from potentially fatal cerebral malaria early in life as the passive immunity acquired in utero wanes. Until recently, however, investigators were unable to document decreased invasion or growth of P. falciparum in red blood cells from thalassemia heterozygotes except under conditions of unusual oxidant stress. Using modified tissue culture conditions, Brockelman and colleagues and, more recently, Pattanapanyasat and colleagues demonstrated decreased parasite multiplication in the red blood cells of β-thalassemia heterozygotes. They theorized that P. falciparum resistance was a consequence of the inability of the parasite to acquire sufficient nutrients from the digestion of hemoglobin in thalassemic red blood cells. In one study, red blood cells with α- and β-thalassemia trait bound greater levels of antibody than did control cells, which could lead to greater removal of parasitized red blood cells and hence provide protection. Recent studies have suggested that parasitized red blood cells from α-thalassemia heterozygotes may have altered membrane properties that more readily allow binding of antibody to the red blood cell and may promote more effective antimalarial immune responses. Erythrocytes from persons with HbH disease also appear to inhibit P. falciparum in vitro. Recently it has been suggested that rosette formation, the binding of uninfected red blood cells to P. falciparum –infected red blood cells, is decreased in persons with thalassemia because of reduced red blood cell size and that such impaired rosette formation may hinder the development of cerebral malaria by lessening sequestration.
The difficulty of documenting the cellular mechanism of P. falciparum resistance in thalassemic erythrocytes in vitro suggests that the heterozygote advantage may be small. The high mortality associated with malaria in endemic regions is a powerful selective force that may be sufficient to amplify a small increase in fitness.
Classes of Mutations That Cause Thalassemia
Thalassemia is the consequence of mutations that diminish (or abolish) production of either the α or β chain of hemoglobin. Molecular cloning, DNA sequencing, and functional analysis of cloned genes have provided the tools with which to dissect the thalassemia syndromes. This analysis has revealed remarkable heterogeneity in the specific alterations in DNA that lead to these clinical syndromes.
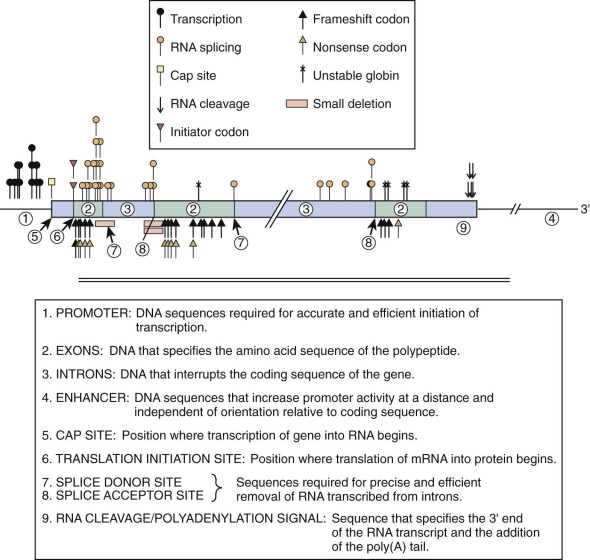
Typically, single nucleotide mutations associated with thalassemia interfere with one of the critical steps in mRNA production ( Fig. 21-6 and Table 21-2 ). Base substitutions alter promoter function, RNA processing, or mRNA translation or modify a codon into a “nonsense codon” that leads to premature termination of translation or substitution of an incorrect amino acid. Insertion or deletion mutations within the coding region of the mRNA create “frameshifts” that prevent the synthesis of a complete, normal globin polypeptide. Large deletions within the α- or β-globin clusters may remove one or more genes and alter regulation of the remaining genes in the cluster. The phenotype that results from the diverse mutations found in thalassemia is determined by the degree of inactivation of the affected gene or genes and the extent of associated increases in expression of other genes within the cluster. In the following section we discuss examples of such mutations. The discussion of various mutations is meant to illustrate different molecular mechanisms that can lead to thalassemia and in no way is meant to be comprehensive. A comprehensive and regularly updated listing of all described mutations can be found at http://globin.cse.psu.edu/ .
| Gene | Position * | Mutation | Classification | Ethnic Group † | Detection ‡ | References |
|---|---|---|---|---|---|---|
| A. Transcription Mutations | ||||||
| β | 1-101 | C→T | β + | Turkish | ||
| Bulgarian | ||||||
| Italian | ||||||
| 2-92 | C→T | β + | Mediterranean | |||
| 3-88 | C→T | β + | American black | (+) Fok I | ||
| Asian Indian | ||||||
| 4-88 | C→A | β + | Kurdish | |||
| 5-87 | C→G | β + | Mediterranean | (−) Avr II | ||
| 6-86 | C→G | β + | Lebanese | |||
| 7-31 | A→G | β + | Japanese | |||
| 8-30 | T→A | β + | Turkish | |||
| Bulgarian | ||||||
| 9-30 | T→C | β + | Chinese | |||
| 10-29 | A→G | β + | American black | (+) Nla III | ||
| Chinese | ||||||
| 11-28 | A→C | β + | Kurdish | |||
| 12-28 | A→G | β + | Chinese | |||
| B. Cap Site Mutation | ||||||
| β | 11 | A→C | β + | Asian Indian | ||
| C. RNA Splicing Mutations | ||||||
| 1. Splice Junction Change in | ||||||
| a . 5′ donor site | ||||||
| α 2 | 1 IVS-1 n. 2-6 | 5-bp deletion | α 0 | Mediterranean | ||
| β | 1 IVS-1 n. 1 | G→A | β 0 | Mediterranean | (−) Bsp M1 | |
| 2 IVS-1 n. 1 | G→T | β 0 | Asian Indian | (−) Bsp M1 | ||
| Chinese | ||||||
| 3 IVS-1 n. 2 | T→G | β 0 | Tunisian | |||
| 4 IVS-1 n. 2 | T→C | β 0 | Black | |||
| 5 IVS-1 5′ end | 44-bp deletion | β 0 | Mediterranean | |||
| 6 IVS-2 n. 1 | G→A | β 0 | Mediterranean | (−) Hph I | ||
| Tunisian | ||||||
| American black | ||||||
| β | 1 IVS-1 n. 130 | G→C | β 0 | Italian | ||
| 2 IVS-1 n. 130 | G→A | β 0 | Egyptian | |||
| 3 IVS-1 3′ end | 17-bp deletion | β 0 | Kuwaiti | |||
| 4 IVS-1 3′ end | 25-bp deletion | β 0 | Asian Indian | |||
| 5 IVS-2 n. 849 | A→G | β 0 | American black | |||
| 6 IVS-2 n. 849 | A→C | β 0 | American black | |||
| 2. Splice Consensus Sequence Change in | ||||||
| a . 5′ donor site | ||||||
| β | 1 IVS-1 n. −3 (codon 29) | C→T | ? | Lebanese | ||
| 2 IVS 1 n. −1 (codon 30) | G→C | Hb Monroe | Tunisian | |||
| American black | ||||||
| 3 IVS 1 n. −1 (codon 30) | G→A | ? | Bulgarian | |||
| 4 IVS-1 n. 5 | G→C | β + | Asian Indian | |||
| Chinese | ||||||
| 5 IVS-1 n. 5 | G→T | β + | Melanesian | |||
| Mediterranean | ||||||
| American black | ||||||
| 6 IVS-1 n. 5 | G→A | β + | Algerian | (+) Eco RV | ||
| Mediterranean | ||||||
| 7 IVS-1 n. 6 | T→C | β + | Mediterranean | (+) Sfa NI | ||
| b . 3′ acceptor site | ||||||
| β | 1 IVS-1 n. 128 | T→G | β + | Saudi Arabian | ||
| 2 IVS-2 n. 843 | T→G | β + | Algerian | |||
| 3 IVS-2 n. 848 | C→A | β + | Iranian | |||
| Egyptian | ||||||
| American black | ||||||
| 3. Mutations within Exons That Affect Processing | ||||||
| β | 1 Codon 19 (Asn-Ser) | A→G | Hb Malay | Malaysian | ||
| 2 Codon 24 (silent) | T→A | β + | American black | |||
| 3 Codon 26 (Glu-Lys) | GvA | Hb E | Southeast Asian | (−) Mnl I | ||
| European | ||||||
| 4 Codon 27 (Ala-Ser) | G→T | Hb Knossos | Mediterranean | |||
| 4. Internal IVS Change | ||||||
| β | 1 IVS-1 n. 110 | G→A | β + | Mediterranean | ||
| 2 IVS-1 n. 116 | T→G | β 0 | Mediterranean | |||
| 3 IVS-2 n. 654 | C→T | β 0 | Chinese | |||
| 4 IVS-2 n. 705 | T→G | β + | Mediterranean | |||
| 5 IVS-2 n. 745 | C→G | β + | Mediterranean | (+) Rsa I | ||
| D. RNA Cleavage And Polyadenylation Mutations | ||||||
| α 2 | 1 Cleavage signal | AATAAA→ AATAAG | α + | Middle Eastern | ||
| Mediterranean | ||||||
| β | 1 Cleavage signal | AATAAA→AACAAA | β + | American black | ||
| 2 Cleavage signal | AATAAA→AATAAG | β + | Kurdish | |||
| 3 Cleavage signal | AATAAA→AATGAA | β + | Mediterranean | |||
| 4 Cleavage signal | AATAAA→AATAGA | β + | Malaysian | |||
| 5 Cleavage signal | AATAAA→A (−AATAA) | β + | Arab | |||
| E. Initiation Consensus Sequence Mutations | ||||||
| α 2 : | 1 Initiation codon | ATG→ACG | α 0 | Mediterranean | (−) Nco I | |
| α 1 : | 2 Initiation codon | ATG→GTG | α 0 | Mediterranean | (−) Nco I | |
| −α: | 3 Initiation codon | ATG→GTG | α 0 | Black | (−) Nco I | |
| −α 3.7II | 4 Initiation consensus | CCACCATGG→CC CATGG | α + | Algerian | ||
| Mediterranean | ||||||
| β | 1 Initiation codon | ATG→AGG | β 0 | Chinese | ||
| 2 Initiation codon | ATG→ACG | β 0 | Yugoslavian | |||
| 3 Initiation codon | ATG→ATA | β 0 | Swedish | |||
| F. Premature Termination Mutations | ||||||
| 1. Substitutions | ||||||
| α 2 | 1 Codon 116 | GAC→TAG | α 0 | Black | ||
| β | 1 Codon 15 | G→A | β 0 | Asian Indian | ||
| 2 Codon 17 | A→T | β 0 | Chinese | (+) Mae I | ||
| 3 Codon 35 | C→A | β 0 | Thai | |||
| 4 Codon 37 | G→A | β 0 | Saudi Arabian | |||
| 5 Codon 39 | C→T | β 0 | Mediterranean | (+) Mae I | ||
| 6 Codon 43 | G→T | β 0 | European | |||
| 7 Codon 61 | A→T | β 0 | Chinese | (−) Hinf I | ||
| Black | ||||||
| 2. Frameshifts | ||||||
| −a | 1 Codons 30/31 | −2 bp (−AG) | α 0 | Black | ||
| β | 1 Codon 1 | −1 bp (−G) | β 0 | Mediterranean | ||
| 2 Codon 5 | −2 bp (−CT) | β 0 | Mediterranean | |||
| 3 Codon 6 | −1 bp (−A) | β 0 | Mediterranean | (−) Cvn I | ||
| American black | ||||||
| 4 Codon 8 | −2 bp (−AA) | β 0 | Turkish | |||
| 5 Codon 8/9 | +1 bp (+G) | β 0 | Asian Indian | |||
| 6 Codon 11 | −1 bp (−T) | β 0 | Mexican | |||
| 7 Codons 14/15 | +1 bp (+G) | β 0 | Chinese | |||
| 8 Codon 16 | −1 bp (−C) | β 0 | Asian Indian | |||
| 9 Codons 27/28 | +1 bp (+C) | β 0 | Chinese | |||
| 10 Codon 35 | −1 bp (−C) | β 0 | Indonesian | |||
| 11 Codons 36/37 | −1 bp (−T) | β 0 | Iranian | |||
| 12 Codon 37 | −1 bp (−G) | β 0 | Kurdish | |||
| 13 Codons 37-39 | −7 bp (−GACCCAG) | β 0 | Turkish | |||
| 14 Codons 41/42 | β 0 | Asian Indian | ||||
| −4 bp (−CTTT) | Chinese | |||||
| 15 Codon 44 | −1 bp (−C) | β 0 | Kurdish | |||
| 16 Codon 47 | +1 bp (+A) | β 0 | Surinamese black | |||
| 17 Codon 64 | −1 bp (−G) | β 0 | Swiss | |||
| 18 Codon 71 | +1 bp (+T) | β 0 | Chinese | |||
| 19 Codons 71/72 | +1 bp (+A) | β 0 | Chinese | |||
| 20 Codon 76 | −1 bp (ϖC) | β 0 | Italian | |||
| 21 Codons 82/83 | −1 bp (−G) | β 0 | Azerbaijani | |||
| 22 Codons 106/107 | +1 bp (+G) | β 0 | American black | |||
| G. Termination Codon Mutations | ||||||
| α 2 | 1 Codon 142 (ter-Gin) | TAA→CAA | Hb Constant Spring | Chinese | ||
| 2 Codon 142 (ter-Lys) | TAA→AAA | Hb Icaria | Mediterranean | |||
| 3 Codon 142 (ter-Ser) | TAA→TCA | Hb Koya Dora | Indian | |||
| 4 Codon 142 (ter-Glu) | TAA→GAA | Hb Seal Rock | Black | |||
| β | 1 Codon 147 (ter-Gin) | Hb Tak | Thai | |||
| H. Unstable Hemoglobin Chains | ||||||
| 1. Amino Acid Substitutions | ||||||
| −α | 1 Codon 14 (Trp-Arg) | Hb Evanston | Black | |||
| α 2 | 2 Codon 109 (Leu-Arg) | T→G | Hb Suan Dok | Southeast Asian | ||
| α | 3 Codon 110 (Ala-Asp) | T→C | Hb Petah Tikvah | Middle Eastern | ||
| α 2 | 4 Codon 125 (Leu-Pro) | Hb Quong Sze | Southeast Asian | |||
| β | 1 Codon 60 | T→A | β + | Italian | ||
| 2 Codon 110 (Leu-Pro) | T→C | Hb Showa-Yakushiji | Japanese | |||
| 3 Codon 112 (Cys-Arg) | Hb Indianapolis | European | ||||
| 4 Codon 127 (Gin-Pro) | Hb Houston | British | ||||
| 5 Codons 127/128 (Gin, Ala-Pro) | −3 bp (−AGG) | β + | Japanese | |||
| 2. Frameshift, Extended Chain | ||||||
| β | 1 Codon 94 | +2 bp (+TG) | Hb Agnana (inclusion body) | Italian | ||
| 2 Codons 109/110 | −1 bp (−G) | Hb Manhattan | Lithuanian | |||
| 3 Codon 114 | −2, +1 (−CT, + G) | Hb Geneva (inclusion body) | French-Swiss | |||
| 4 Codons 128-135 | Net-10 bp | |||||
| β + (inclusion body) | Irish | |||||
| 3. Premature Termination | ||||||
| β | 1 Codon 121 | G-T | β 0 (inclusion body) | Greek-Polish | ||
| French-Swiss | ||||||
| British | ||||||
* The position specifies the location in the gene at which the point mutation occurs. Positions are specified with reference to the start site for transcription (Cap site), the position within the intron (IVS), or the position of the codon.
† When more than one ethnic group is indicated, the mutation has had more than one origin.
‡ Loss (−) or gain (+) of a restriction enzyme site with mutation is indicated; the remainder of the mutations can be detected with allele-specific oligonucleotides (see the section on direct detection of thalassemia mutations).
Mutations Affecting Gene Transcription
Point mutations within promoter sequences recognized by transcription factors tend to reduce the affinity with which these proteins bind and typically leads to reduced gene transcription. Analysis of the promoter for the β-globin gene in patients with β-thalassemia has identified a variety of mutations clustered in the ATA and CACC motifs ( Fig. 21-7 ; see also Table 21-2 ). These mutations are associated with preservation of some β-globin expression and hence are customarily associated with the phenotype of thalassemia intermedia. The C→T substitution at position –101, which results in a particularly mild defect, is associated with the “silent carrier” phenotype in heterozygous carriers. Although the CCAAT box is highly conserved in globin genes, no mutations within this motif have been identified in thalassemia. Rare mutations in transcription factors that result in thalassemia have been detected, and exceedingly rare families have been identified in which a thalassemia mutation is unlinked to the globin clusters (see the section on mutations not linked to the globin gene clusters that alter globin gene expression ).
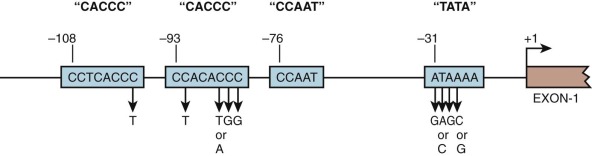
Mutations of the ATA box presumably reduce binding of TBP and therefore lead to decreased transcription initiation. Substitutions in the CACC motifs decrease the affinity of binding by several transcription factors, including the erythroid-specific factor EKLF/KLF1 and the ubiquitous protein Sp1. Studies showing that mice engineered to lack EKLF experience lethal β-thalassemia at the fetal liver stage have established EKLF as a β-globin activator protein in vivo, and patients with mutations in EKLF have reduced production of the adult β-globin gene. Human β-thalassemias resulting from mutation of a single CACC motif are presumably mild because of the presence of one normal CACC motif within the promoter.
In addition to the protein-binding sites in the promoter, proper transcription depends on sequences surrounding the start site of transcription (known as +1). These sequences often display functional activity in in vitro assays and herald the binding of specific protein complexes to this type of element, termed the Inr . Mild β-thalassemia has been associated with a base substitution (A→C) at +1. This substitution has been shown to impair the β-globin Inr. The proteins that mediate this effect are unknown.
A novel mechanism by which transcription at the α-globin locus can be disrupted has been described and serves as a paradigm for a unique class of mutations that can cause human disease. Higgs and colleagues found a variant single nucleotide change upstream of the α-globin genes that creates a binding site for the transcription factor GATA1, which in turn produces a novel promoter that competes with the endogenous α-globin promoters for interaction with upstream enhancer elements such as HS-40. As a result of this mutation, α-globin gene synthesis is reduced and α-thalassemia results. This mutation is present in approximately 4% of the Melanesian population.
RNA Processing Defects in Thalassemia
The importance of RNA splicing for the formation of functional mRNA cannot be overemphasized. As discussed earlier, removal of introns must be precise to the nucleotide for a continuous, translatable mRNA to be generated from an mRNA precursor. As soon as introns were discovered, it was hypothesized that mutations affecting RNA splicing would probably be involved in the thalassemia syndromes. Apart from its role in constructing a functional mRNA, RNA splicing also appears to be a determinant of mRNA stability and is possibly coupled to RNA transport from the nucleus to the cytoplasm.
Mutations That Alter Splice Junctions or Splice Consensus Sequences
Mutations at the 5′ donor site (GT) or at the 3′ splice acceptor site (AG) abolish proper splicing of the pre-mRNA transcript and result in α 0 – or β 0 -thalassemia ( Fig. 21-8 ; see also Table 21-2 ). Substitutions at other sites within the splice junction consensus sequence have varied effects; because some correctly spliced RNA is produced, albeit a reduced amount, a β + -thalassemia phenotype ensues.
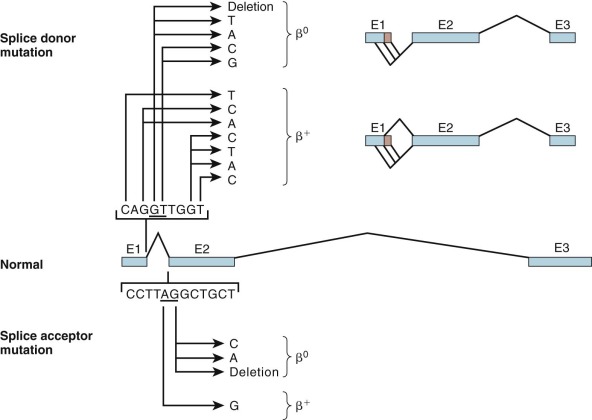
Mutations within the splice site or the splice site consensus sequences favor improper processing of the mRNA precursor. These secondary splicing events, which are not seen under normal circumstances, occur at positions that resemble splice site consensus sequences. Splicing at these “cryptic” sites generates aberrantly processed, nonfunctional globin mRNA (see Fig. 21-8 ). Mutations within the β-globin IVS-1 splice donor site activate two cryptic donor sites in exon 1 and a third site in IVS-1, whereas mutation in the IVS-2 splice donor activates a cryptic donor site in IVS-2. Mutation of the IVS-2 splice acceptor site activates an upstream cryptic splice acceptor at position 579 in IVS-2. These incorrectly spliced mRNA molecules sustain either insertions or deletions in the coding region and also shifts in the translational reading frame downstream of the cryptic splice site. The polypeptide synthesized beyond this point bears no resemblance to the globin chain and is often prematurely shortened by a termination codon encountered in the new reading frame.
Mutations within Exons That Create an Alternative Splice Site
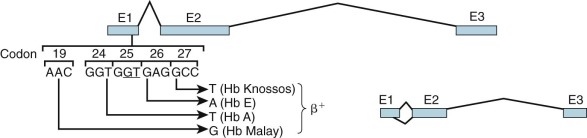
RNA from β-thalassemia genes with mutations in the IVS-1 donor splice site may be processed via a cryptic splice donor site GTG GT GAGG in exon 1 (codons 24 through 27). Four independent mutations have been identified that activate this cryptic site in the presence of a normal IVS-1 splice donor site ( Fig. 21-9 ; see also Table 21-2 ). These mutations appear to enhance the ability of the cryptic site to compete with the normal site for binding of the splicing complex. A T→A mutation at codon 24 is “silent” at the translational level, yet approximately 80% of RNA transcripts are spliced at this incorrect site; hence, mild β + -thalassemia ensues. Two mutations—GAG→AAG in codon 26 and GCC→TCC in codon 27—lead to amino acid replacements that produce the hemoglobin variants HbE and Hb Knossos, respectively, in normally processed mRNA. Because a proportion of transcripts are aberrantly spliced, mild β + -thalassemia results. An analogous mutation in codon 19 produces β + -thalassemia with the hemoglobin variant Hb Malay, representing mutations that lead to thalassemic hemoglobinopathies.
Mutations within Introns That Create an Alternate Splice Site
Mutations within β-globin IVS-1 may create a new splice acceptor sequence ( Fig. 21-10 ; see also Table 21-2 ). In the first of this class of mutations to be characterized, a G→A substitution at position 110 (19 nucleotides upstream of the normal intron/exon boundary), the majority of globin mRNA precursors are spliced at this alternate site. Because the incorrectly spliced mRNA contains 19 nucleotides from IVS-1, a shift in the reading frame leads to premature termination of translation. A T→G mutation at position 116 of IVS-1 creates a new acceptor site that is used exclusively, thereby leading to little or no normal β-globin mRNA production and to β 0 -thalassemia.
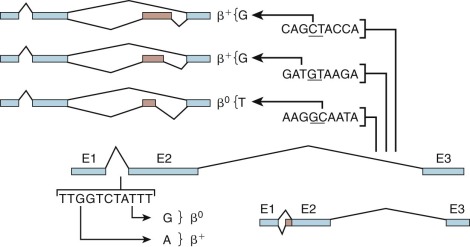
Three mutations in IVS-2 create new donor sites and activate an upstream cryptic donor site located 579 nucleotides from the exon 2–IVS-2 boundary (see Table 21-2 and Fig. 21-10 ). The consequence of these mutations is the insertion of a fourth “exon” derived from sequences within IVS-2. Although the normal donor and acceptor sites are unaffected, little or no correctly spliced β-globin mRNA may be produced.
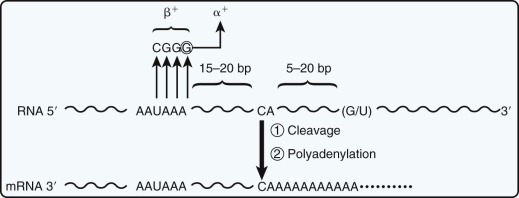
RNA Cleavage and Polyadenylation Defects
Proper cleavage at the 3′ end of the pre-mRNA and subsequent poly(A) addition depend on the integrity of the AAUAAA signal in the 3′-untranslated region. The importance of the polyadenylation signal for efficient production of globin mRNA was first demonstrated in α-thalassemia. An AAUAAA→AAUAAG mutation in the α 2 gene reduces the efficiency of cleavage-polyadenylation of precursor RNA and leads to “run-on” transcripts that terminate downstream of the gene (see Fig. 21-12 ). Mutations in the AAUAAA element have also been described in β-thalassemia, in which the presence of elongated in vivo transcripts has been demonstrated. The transcripts appear to terminate at the next AAUAAA signal, which is present approximately 900 nucleotides downstream of the normal cleavage site. These mutations lead to a moderate reduction in the level of β-globin mRNA and a β + phenotype.
Mutations Affecting mRNA Translation Initiation
Translation begins at an AUG codon that usually lies within the consensus sequence (GCC)GCC(A/G)CC AT GG. Substitutions within the AUG codon abolish translation, whereas those in other positions of the consensus often result in less efficient initiation of translation.
Four mutations in α-globin genes alter the consensus sequence and impair translation (see Table 21-2 ). Three of them affect the AUG Inr. No globin polypeptide is produced because the next downstream Inr is in a different reading frame. The fourth α-globin mutation in this class, found on a chromosome in which one α-globin gene was deleted, alters the consensus sequence by the deletion of two bp and reduces mRNA translation to 50% of normal. Two AUG Inr mutations of the β-globin gene have been described, and both are of the β 0 type (see Table 21-2 ).
Premature Termination (Nonsense) Mutations
Nucleotide substitutions within the coding region are innocuous if they occur in the third position of a codon and do not alter the amino acid inserted during translation. Substitutions that alter codons from one amino acid to another lead to hemoglobin structural variants. Some substitutions change a triplet coding for an amino acid to a stop codon (UAG, UUA, or UGA). Such chain termination (or nonsense) mutations abort mRNA translation and lead to the synthesis of a truncated polypeptide. Moreover, nonsense mutations also reduce the amount of stable mRNA generated, which is a reflection of coupling between mRNA biogenesis and mRNA translation ( Fig. 21-11 ).
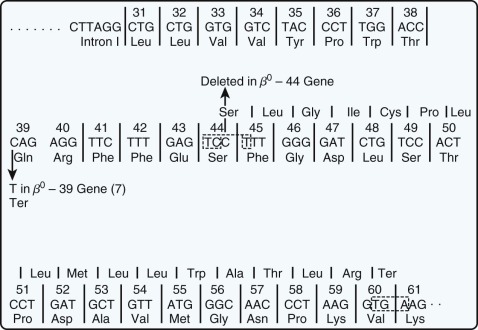
Chang and Kan described the first nonsense mutation in β-thalassemia in which a lysine codon at amino acid position 17 was converted to a stop codon (AAG→UAG). Although no β-globin chains were produced in vivo, complete translation of the abnormal mRNA could be achieved in a cell-free extract capable of protein synthesis by the addition of a “suppressor” transfer RNA (tRNA) that inserts a serine at the UAG codon. Several other nonsense mutations causing thalassemia have been described ( Fig. 21-12 ; see also Table 21-2 ). In addition, single or dinucleotide insertions or deletions have been observed that alter the translational reading frame and introduce a premature stop codon as a consequence. Two termination mutations have been described in the α-globin genes, one that introduces a stop codon and the other a frameshift. In addition, frameshift mutations have been described that result in abnormal elongation of globin chains (see the section on unstable β-globin chains ).
mRNA molecules with termination mutations often do not accumulate to a normal level in vivo. The extent of this effect is variable and depends on the specific mutations; deletion of the third nucleotide (C) from codon 41 ( Fig. 21-13 ) leads to complete absence of globin mRNA, whereas a single substitution in the β39 codon allows the accumulation of roughly 5% to 10% of the normal amount of globin mRNA. The basis for the quantitative deficiency in these mRNA species is of considerable interest. Some data suggest that such mutations lead to intranuclear degradation of abnormal globin RNA and suggest a link between mRNA translation and nuclear RNA processing or nuclear to cytoplasmic transport of mRNA. Experimental studies in tissue culture systems have shown that the deficiency in β-globin mRNA accumulation is specific for nonsense mutations and is not observed with missense mutations ; a suppressor tRNA that allows the abnormal mRNA molecule to be translated completely will correct the quantitative deficiency in globin mRNA. Recent work from a number of investigators has begun to unravel the molecular machinery that mediates this phenomenon, which has been termed nonsense-mediated decay , and appears to play an important role in normal physiology, as well as pathologic states.
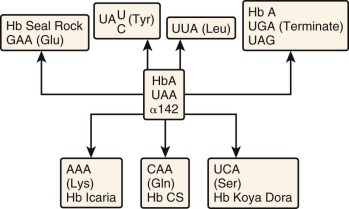
Termination Codon Mutations
UAA is the normal termination codon for both α- and β-globin mRNA translation. The 3′-untranslated regions are 109 and 132 nucleotides for α- and β-globin mRNA, respectively. A single nucleotide substitution in the termination codon could either create another stop codon (UAG) or permit incorporation of an amino acid at this position and translation of the otherwise untranslated 3′ sequences until the next in-frame stop codon. Four termination codon mutations involving the α 2 gene have been reported (see Table 21-2 ). These mutants differ only in the specific amino acid incorporated at the terminator codon position (see Fig. 21-13 ). Translation terminates in each instance at a UAA codon in the polyadenylation signal (AA UAA A) downstream, and a 172–amino acid polypeptide is produced. The first of these elongated α chains to be described was found in Hb Constant Spring (CS). The α chain in this hemoglobin has a glycine substituted at codon 142. Hb CS produces an associated thalassemia phenotype because of a marked reduction in α 2 -globin mRNA stability.
Mutations that give rise to elongated β-globin chains have also been described. Hb Tak is a 157–amino acid product of a β-globin mRNA molecule containing two inserted nucleotides in the terminator codon 147. An analogous elongated β-globin with 157 amino acids found in Hb Cranston reflects a dinucleotide insertion in codon 147, but red blood cells containing Hb Cranston are morphologically normal. The mechanism by which the β Tak mutation causes thalassemia has not been elucidated.
Mutations Affecting Globin Chain Stability
Hemoglobin Assembly.
Shortly after synthesis is completed, α- and β-globin chains bind a heme moiety and rapidly associate into α 1 β 1 dimers in a noncovalent reaction that is nearly irreversible under physiologic conditions. The majority of the heme contact points are present in the portion of the globin chains encoded by exon 2, whereas most α 1 β 1 contacts are located within the exon 3 domain ( Fig. 21-14 ). These dimers may then reversibly associate with other dimers to form the hemoglobin tetramer. Formation of the α 1 β 1 dimer, therefore, is the principal controlling step in the assembly of hemoglobin. This subject is discussed in greater depth in Chapter 19 .
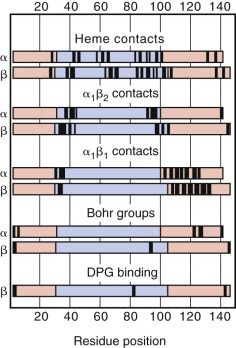
Hemoglobin assembly is an important determinant of the final hemoglobin composition of the erythrocyte. The rate constant of dimer formation depends greatly on the surface electrostatic charge of the subunits. α-Globin has a net positive surface charge, whereas β-globin has a net negative surface charge. The other normal β-like globin chains, γ-globin and δ-globin, dimerize with the α chain at a lower rate. δ-Globin has a lower net negative surface charge than does β-globin, and the significant structural differences between γ- and β-globin presumably account for the differing dimerization rates. When β-globin chains are in limited supply (i.e., β-thalassemia), HbA 2 and HbF levels may increase because of enhanced dimerization with α chains, independent of changes in the production of δ and γ chains. In α-thalassemia or iron deficiency, HbA 2 and HbF levels will decrease because of competition with β chains for the limited number of α chains. Similarly, β-chain variants may have decreased (β S and β E ) or increased (β Baltimore ) affinity for α chains based on their net surface charge. The net hemoglobin composition of the cell is determined by these simple rules of competition based on the relative affinity of hemoglobin subunits.
An efficient, energy-dependent protein quality control system is present in erythrocytes that rapidly degrades free globin chains while leaving chains that are incorporated into dimers or tetramers unaffected. Changes in globin chain structure that result from amino acid substitutions, premature chain termination, or chain elongation may slow or block the formation of stable α 1 β 1 dimers and lead to rapid degradation of the globin chain. In some instances, as discussed later, mutations may also enhance association of the free globin chain with the cell membrane and thereby promote oxidative damage to the membrane and shortened red blood cell survival.
An erythroid-specific α-hemoglobin–stabilizing protein (AHSP) acts as a chaperone for free α-globin chains. In the absence of this protein, mice exhibit mild hemolytic anemia as a result of α-globin precipitation. When this protein is absent in the presence of a β-thalassemia mutation in mice, a much more severe thalassemic phenotype occurs. However, it is currently unclear whether variation in the structure or expression of this gene in humans causes phenotypic variability in patients with β-thalassemia. One study has suggested that this was not the case in patients with HbE–β-thalassemia in Thailand. Another study has suggested that expression levels of this gene vary in humans and that such levels may correlate with the severity of β-thalassemia. Further studies will need to be conducted to better understand whether AHSP plays a role as a modifier gene in β + -thalassemia.
During hemoglobin biosynthesis it is also important that the globin chains not be produced in excess of heme. A protein called heme-regulated inhibitor (HRI) kinase appears to mediate this function by inhibiting globin chain translation when an erythroid precursor is deficient in heme. When HRI is absent in mice with a β-thalassemia mutation, the thalassemic phenotype is exacerbated and lethality of the mice results.
Unstable α-Globin Chains.
One such unstable variant was identified on sequencing of a mutant α-globin gene (see Table 21-2 ). The α Quong Sze variant, which contains a Leu→Pro substitution at position 125, is so unstable that the mutant globin chain cannot be detected by biosynthetic studies in intact cells or by conventional hemoglobin electrophoresis. The α Quong Sze chain appears to be stable once it is incorporated into the hemoglobin tetramer. Three other similar unstable α-globin chain mutations have been described (see Table 21-2 ).
Unstable β-Globin Chains
Several unstable β-globin chains have been associated with thalassemia (see Table 21-2 ). Five mutations lead to amino acid substitutions in the β-globin chain. Frameshift mutations in the third exon result in the synthesis of an elongated β-globin chain with a novel carboxy-terminal. A premature termination mutation in exon 3 has also been described. Many of the unstable β-globin chain mutations in exon 3 are associated with a dominantly inherited form of thalassemia (see the section on dominant β-thalassemia ). Thein and colleagues proposed that alterations in β-globin structure in exon 3 interfere with α 1 β 1 dimer formation yet may permit binding of heme to the mutant globin chain through contacts in the exon 2 domain. These free, heme-associated β chains may be more resistant to proteolysis and associate with the cell membrane, where they form “inclusion bodies” and induce oxidative damage.
Thalassemic Hemoglobinopathies
The mutations described in this section, taken together with the RNA-processing mutants HbE, Hb Knossos, and Hb Malay and the termination mutant Hb Tak, include a distinctive set characterized by structural changes in the hemoglobin molecule and a thalassemia phenotype. These mutations are often referred to as “thalassemic hemoglobinopathies” (see the section on thalassemic hemoglobinopathies ) and are characterized clinically by a syndrome of ineffective erythropoiesis. Other globin variants may be associated with mild hypochromia, microcytosis, and chronic hemolysis because of instability and degradation of hemoglobin tetramers and are discussed in detail in Chapter 19 . Because thalassemia is the consequence of an imbalance in α- and β-globin chains, these variants are not considered part of the spectrum of thalassemia.
Identification, Characterization, and Ethnic Distribution of β-Thalassemia Mutations
The hemoglobin disorders serve as a paradigm for the molecular analysis of genetic disease. Study of β-thalassemia was aided by the introduction of methods for identifying and characterizing mutant alleles that are now widely used. Accordingly, the identification of β-thalassemia mutations in many ethnic groups is nearly complete.
In this section we briefly outline molecular techniques that have been applied to the characterization of β-thalassemia mutations. An understanding of these methods is important not only because they are broadly used in the study of other genetic diseases but also because they are directly relevant to strategies for genetic screening and prenatal diagnosis of β-thalassemia.
Haplotype Analysis
The first several β-thalassemia mutations were identified by cloning and sequencing β-globin genes isolated from persons with β-thalassemia major. Because certain mutations are extremely common, a nondirected strategy is inefficient because β genes with common mutations will be repeatedly studied. For example, 95% of the β-thalassemia alleles on the island of Sardinia contain the codon 39 nonsense mutation.
To facilitate the search for new β-thalassemia mutations, Orkin, Kazazian, Kan, and others introduced the concept of haplotype analysis to the study of thalassemia. Naturally occurring, genetically neutral, nonselected sequence differences among individuals constitute polymorphisms, which are estimated to occur roughly once every 100 bp. These sequence differences are heritable, and those residing close to one another on a chromosome tend to be inherited together, a property known as linkage . A subset of polymorphisms will alter the cleavage site for a restriction enzyme or create a site where one did not exist. Therefore, when DNA from unrelated individuals is digested with a restriction enzyme and analyzed by Southern blotting, polymorphisms in the restriction enzyme digest pattern may be observed and are referred to as restriction fragment length polymorphisms (RFLPs). Within the 60 kb of the human β-globin cluster, more than such 20 RFLPs are known ; at least 13 have been identified in the α cluster. The pattern of these RFLPs (each based on the presence [+] or absence [−] of a restriction enzyme cutting site) along the chromosome defines a haplotype of associated or linked polymorphisms. Seven RFLPs were used initially to define nine distinct haplotypes (I through IX) of the β-gene cluster in the analysis of thalassemia mutations in Greek and Italian populations from the Mediterranean basin ( Fig. 21-15 ).
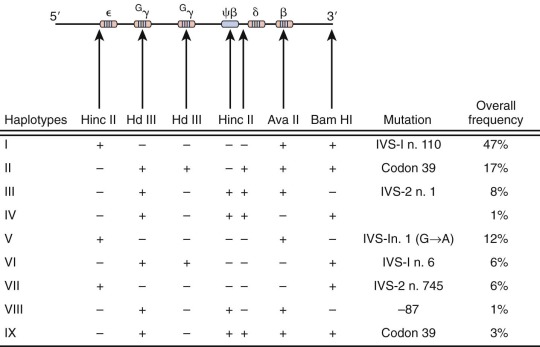
Close inspection of these haplotypes revealed a nonrandom association of restriction digest patterns within the β-gene cluster ( Fig. 21-16 ). The pattern of restriction sites 5′ of the δ-globin gene is inherited as a group, whereas restriction sites downstream (including the β-globin gene) track as another set. In all populations only a few haplotypes predominate. The full spectrum of haplotypes is derived from random association over evolutionary time between the 5′ and the 3′ subhaplotypes, presumably reflecting the presence of a recombination “hot spot” lying between these regions.
The generation of haplotypes appears to be an ancient event predating racial dispersion. Consequently, a specific haplotype may be found in diverse ethnic and racial groups from different geographic locations. The introduction of malaria as a selective pressure for certain random mutations is a more recent phenomenon. A mutation leading to thalassemia would be under positive selection and amplified within a population; accordingly, the mutation resides on the haplotype background in which it originated in that ethnic group. Several conclusions were derived from the study of different racial groups. Within a single population both normal and thalassemia β-globin genes are found on the same haplotype, but specific thalassemia mutations tend to be linked to a single haplotype. Individual thalassemia mutations are generally restricted to a single population (see Table 21-2 ). In circumstances in which specific mutations are found in different populations, the identical thalassemia mutation may have occurred and been selected independently and will be found on a different haplotype background. This observation has provided a sound genetic basis for the belief that thalassemia has had multiple distinct origins throughout the world. In circumstances in which a specific mutation is found on more than one haplotype within a population, the 3′ subhaplotypes (where the β-globin gene resides) may be identical, whereas the 5′ subhaplotypes differ because of recombination between the two subhaplotypes (for an example, see codon 39 mutation, see Fig. 21-15 ). By this mechanism, specific mutations can be distributed to new haplotypes within an ethnic group, and an independent origin of the mutation need not be invoked.
New thalassemia mutations were identified by cloning thalassemia β-globin genes from distinct haplotypes within a population, thereby avoiding the likelihood of repeated cloning of the same common mutation. In this way, the great diversity of thalassemia mutations was elucidated.
Ethnic Distribution
Determining the incidence of specific β-thalassemia mutations in different ethnic groups is particularly relevant to strategies for prenatal diagnosis of or preconception screening for thalassemia. Nearly complete surveys of thalassemia mutations have been performed in Greek and Italian, Asian Indian, American black, Sardinian, Chinese, Lebanese, Turkish, Spanish, Sicilian, Thai, Kurdish Jewish, and Japanese populations. From these studies, several general conclusions can be drawn. First, in each ethnic group a small subset of mutations (as few as four or six) constitute more than 90% of the mutant alleles. This characteristic is particularly striking on the island of Sardinia, where the codon 39 nonsense mutation accounts for 95% of the β-thalassemia genes and a codon 6 frameshift represents another 4%. Second, the remaining 5% to 10% of mutant alleles in an ethic group are divided among a larger number of rarer alleles. For example, four alleles account for 90% of the β-thalassemia genes in Chinese, and 11 rare alleles account for the remaining 10%. Third, several mutations appear to have originated independently in different ethnic groups and are present on different haplotype backgrounds, as discussed earlier. For example, the IVS-2 No. 1 (G→A) mutation is present in Mediterranean, Tunisian, and American black populations, whereas the IVS-1 No. 5 (G→C) substitution is present in Asian Indians, Chinese, and Melanesians. Finally, as a consequence of the large number of mutations present in each population, most persons with severe β-thalassemia are genetic compound heterozygotes for two different thalassemia mutations.
Mutations That Affect β-Globin Gene Regulation
Deletions within the β-globin gene cluster often lead to thalassemia. Many of these deletions are associated with a significant increase in the level of HbF, a finding that distinguishes them from the common varieties of β-thalassemia. Ordinarily, heterozygous carriers of β-thalassemia have an increase in the level of HbA 2 (to >3% of total hemoglobin) and, at most, a slight increase in the level of HbF. Before detailed molecular analysis was available, these conditions were broadly grouped into two categories, hereditary persistence of fetal hemoglobin (HPFH) and δβ-thalassemia ( Table 21-3 ). HPFH heterozygotes have normocytic, normochromic red blood cells, whereas δβ-thalassemia heterozygotes have hypochromic and microcytic cells. Many HPFH heterozygotes have high levels of HbF (up to or greater than 30%) with a uniform or pancellular distribution in circulating erythrocytes, whereas in δβ-thalassemia heterozygotes, the amount of HbF is less abundant and it is present in an uneven or heterocellular distribution among red blood cells. In rare persons who are homozygous for either condition, only HbF is found. HPFH homozygotes have normal or slightly elevated total hemoglobin concentrations, their red blood cells are slightly hypochromic and microcytic, and globin synthesis is modestly imbalanced. Thus these mutations are appropriately considered along with the thalassemia mutations.
| HPFH | δβ-Thalassemia | |
|---|---|---|
| Red blood cell morphology | Normal | Abnormal |
| MCH | Nearly normal | Decreased |
| Hematocrit | Normal | Slightly decreased |
| HbF (%) | 15-30 | 1-15 |
| HbF distribution in red blood cells | Pancellular | Heterocellular |
Deletion mutations of the β-globin gene cluster represent in vivo experiments of nature useful in developing and validating experimental models of gene regulation. Multiple regulatory elements are present within the cluster, and the clinical phenotypes observed with specific deletions relate to removal of one or more such regulatory elements. More than 30 deletion mutations have been described ( Table 21-4 and Fig. 21-17 ) ; they vary greatly in size from a few hundred base pairs of the β-globin gene to more than 100 kb with loss of the entire cluster. Recent studies have shown how insight from such deletions and mapping regions that diverge between the HPFH and δβ-thalassemia deletions can help to elucidate key regulatory regions important for HbF expression. For example, a region upstream of the δ-globin gene has been implicated in playing a key role in silencing the γ-globin genes and binds the HbF silencing factor BCL11A. In addition to deletion mutations, significant elevations of HbF in adults may arise as a result of single base substitutions within the γ-globin gene promoters ( Table 21-5 ). Such mutations, also classified as nondeletion HPFH, appear to enable the γ-globin genes to “capture” the influence of the LCR at the adult stage. Persons with these HPFH mutations have HbF values ranging from only slightly elevated to more than 20%, which can be distributed in either a heterocellular or pancellular fashion.
| Type | Ethnic Group | Deletion Size (kb) | Deletion Coordinates | HbF Level in Heterozygotes (%) | Other Information | References |
|---|---|---|---|---|---|---|
| A. A γ 0 | ||||||
| 1 | 2.5 | 37.7-40.2 | 0.2 | “Silent” | ||
| B. δ 0 | ||||||
| 1 | Corfu | 7.20 | 48.9-56.1 | 1.1-1.6 | δ 0 -Thalassemia | |
| C. β 0 | ||||||
| 1 | Indian | 0.619 | 63.?-64.0 | Normal | β 0 -Thalassemia | |
| 2 | American black | 1.393 | 61.6-63.0 | 7.0-7.9 | β 0 -Thalassemia | |
| 3 | Dutch | 12.6 | 59.7-72.3 | 4-11 | β 0 -Thalassemia | |
| 4 | Turkish | 0.29 | 62.1-62.4 | 2.7-3.3 | β 0 -Thalassemia | |
| Jordanian | 9.4 | β 0 -Thalassemia | ||||
| 5 | Czech | 4.237 | 58.9-63.1 | 3.3-5.7 | β 0 -Thalassemia | |
| Canadian | ||||||
| D. (δβ) 0 | ||||||
| 1 | Sicilian | 13.377 | 56.0-69.4 | 5-15 | ||
| 2 | Spanish | ≅114 | 52.2-? | 5-15 | ||
| 3 | American black | 12.0 | 52.3-64.1 | 25 | Pancellular * | |
| 4 | Japanese | >130 | 43.1-? | 5-7 | ||
| 5 | Laotian | 12.5 | 56.0-68.5 | 11.5 | ||
| 6 | Macedonian | 18-23 | 54-74 | 7-14 | ||
| 7 | Mediterranean | 7.4 | 55.3-62.7 | 1-5 | Hb Lepore | |
| E. G γ + (Aγδβ) 0 | ||||||
| 1 | Indian | Total 8.3 kb | 40.1-40.9 | 10-15 | ||
| Iranian | Deleted | 55.5-63.0 | ||||
| Kuwaiti | 14.6 kb inverted | 40.9-55.5 | ||||
| 2 | American black | 35.7 | 40.7-76.4 | 6-16 | ||
| 3 | Turkish | 36.22 | 37.1-73.3 | 10-15 | ||
| American black | ||||||
| 4 | Malaysian (1) | >27 | 37.0-? | Unknown | ||
| 5 | Malaysian (2) | >40 | 39.1-? | Unknown | ||
| 6 | Chinese | ≅100 | 40.5-? | 10-15 | ||
| 7 | German | 53.0 | 37.6-90.6 | 9.9-12.5 | ||
| 8 | Cantonese | >43 | 37.0-? | 20 | ||
| F. (γδβ) 0 | ||||||
| 1 | Hispanic | 39.5 | −19.5-10.0 | |||
| 2 | English | >100 | ?-35.9 | |||
| 3 | Dutch | 99.4 | ?-59.8 | |||
| 4 | Anglo-Saxon | 95.9 | ?-62.6 | |||
| 5 | Mexican | >105 | ?-? | |||
| 6 | Scotch-Irish | >105 | ?-64-71 | |||
| 7 | Yugoslavian | >148 | ?-? | |||
| 8 | Canadian | >185 | ?-? | |||
| G. HPFH | ||||||
| 1 | American black | ≅106 | 51.2-? | 20-30 (5% G) † | HPFH-1 | |
| 2 | Black (Ghana) | ≅105 | 47-? | 20-30 (30% G) † | HPFH-2 | |
| 3 | Indian | 48.5 | 45.0-93.5 | 22-23 (70% G) † | HPFH-3 | |
| 4 | Italian | 40.0 | 50.0-90.0 | 14-30 | HPFH-4 | |
| 5 | Italian | 12.9 | 51.6-64.5 | 16-20 (15% G) † | ||
| 6 | Kenyan | 22.8 | 40.0-62.8 | 5-8 | Hb Kenya | |
* In a compound δβ-thalassemia/HbS heterozygote.
† Percentage of G γ in total γ (Gγ/Gγ + Aγ). Forty percent G γ is the normal value for an adult.
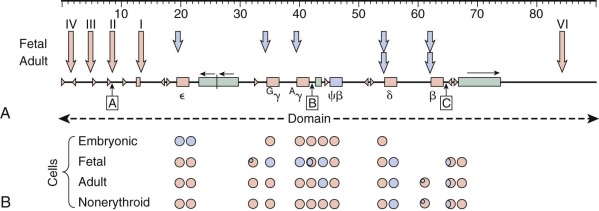
| Type | Molecular Defect | γ Gene | Ethnic Group | Percentage of HbF in Heterozygotes | Chains G γ, A γ | Distribution of HbF | References |
|---|---|---|---|---|---|---|---|
| 1 | C→G at −202 | G | Black | 15-20 | G only | Pancellular | |
| 2 | C→T at −202 | A | Black | 2-3 | 93% A | Heterocellular | |
| 3 | +C insertion at −200 | G | Tunisian | 18-27 | G only | ||
| 4 | T→C at −198 | A | British * | 4-12 | 90% A | Heterocellular | |
| 5 | C→T at −196 | A | Chinese | 10-15 | 90% A | Heterocellular † | |
| Italian ‡ | 10-20 | 95% A | Pancellular | ||||
| 7 | T→C at −175 | G | Italian | 20-30 | 90% G | ||
| Black | 30 † § | G only | Pancellular | ||||
| 8 | T→C at −175 | A | Black | 35-40 | 80% A | Pancellular | |
| 9 | G→A at −161 | G | Black | 1-2 | Heterocellular | ||
| 10 | G→T at −158 ‖ | G | Saudi ¶ | 2-4 | G > A | Heterocellular | |
| 11 | G→A at −117 | A | Mediterranean # | 10-20 | 95% A | Pancellular | |
| Black | 10-20 | 85% A | Pancellular | ||||
| 12 | C→G at −114 | G | Australian | 8.5 | 90% G | ||
| 13 | C→T at −114 | G | Japanese | 11-14 | G > A | ||
| 14 | C→T at −114 | A | Black | 3-5 | 90% A | ||
| 15 | Deletion −114 to −102 | A | Black | 30 † § | 85% A ‡ | ||
| 16 | X-Linked | G | “Swiss” ** | 0.8-3.4 | Heterocellular | ||
| 17 | Unknown † | G | German | 5-8 | A and G | Heterocellular | |
| 18 | Unknown | G | “Georgia” | 2.6-6 | G > A | Heterocellular | |
| 19 | Unknown | G | “Seattle” | 3.7-7.8 | G = A | Heterocellular |
* Also found in whites in North America.
† More than 80% of the red blood cells contain HbF.
‡ Also found in cis to codon 39 mutation (Sardinian δβ-thalassemia).
‖ This determinant appears to increase HbF only in patients with erythropoietic stress.
¶ Identified in Greek and Sardinian families.
# Also found in several other ethnic groups.
HPFH and δγ-thalassemia mutations are uncommon, and persons who inherit these mutations are asymptomatic or have mild disease. Their importance relates to the insight that they provide into globin gene regulation and the role of HbF in modulating disease severity in patients with severe γ-thalassemia or sickle cell anemia.
Isolated point mutations of the δ-globin gene, similar to those in β-thalassemia, may lead to “δ-thalassemia.” δ-Thalassemia is a benign condition with no clinical significance that, when inherited with a β-thalassemia mutation, may lead to a normal or low HbA 2 thalassemia phenotype in heterozygotes.
Crossover Globins: Hemoglobin Lepore and Hemoglobin Kenya
Deletion mutations in the α- and β-globin gene clusters arise through unequal homologous recombination or through nonhomologous (illegitimate) recombination. In contrast to the α-globin cluster, in which there are blocks of tandem duplicated sequences (discussed later), the only directly repeated homologous segments of DNA in the β cluster are the globin genes themselves. Hence mutations arising from homologous but unequal crossover in the β-globin cluster are relatively uncommon and usually involve two globin genes directly ( Fig. 21-18 ). Two crossover hemoglobins, Hb Lepore and Hb Kenya, are associated with thalassemia.
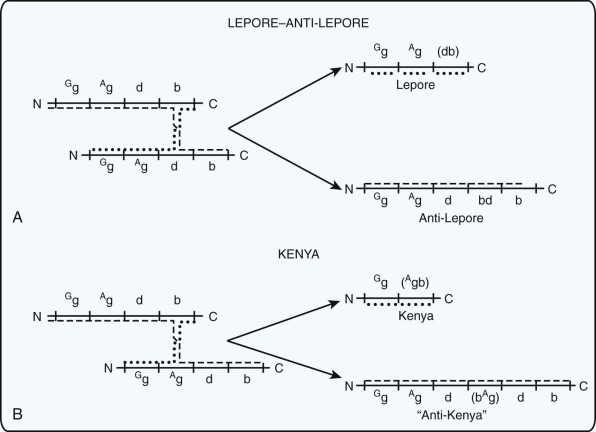
In 1958, Gerald and Diamond identified a minor hemoglobin component by starch gel electrophoresis in the blood of parents of a patient with thalassemia major. Structural analysis revealed that the hemoglobin was composed of α-globin and a heretofore undescribed globin consisting of fusion of the 80 to 100 amino-terminal amino acids of δ-globin with the carboxyl portion of β-globin. This “chimeric” globin polypeptide, named Lepore after the family in which it was first described, arose from an unequal, homologous recombination event between the δ-globin and β-globin genes ( Fig. 21-18 ). It is poorly expressed because transcription of the fusion gene is under control of the δ-gene promoter. Other Lepore-type globins have been characterized in which the relative contribution of the δ and β genes to the fusion protein varies as a result of a different point of crossover. Homozygotes for Hb Lepore have 90% HbF and approximately 10% Hb Lepore, but no HbA or HbA 2 , whereas heterozygotes have mainly HbA, 2% to 4% Hb Lepore, and 3% to 5% HbF. In heterozygotes, red blood cells have a very heterogeneous HbF content. Anti-Lepore globins with the amino-terminal sequence of β-globin and the carboxyl sequence of δ-globin have also been described (see Fig. 21-18 ). Persons with anti-Lepore genes also have two normal δ- and two normal β-globin genes; consequently, their red blood cells lack any stigmata of thalassemia. The anti-Lepore globins are produced in very low amounts, perhaps because of sequences within the large intron of the δ-globin gene that may reduce mRNA production.
Hb Kenya, another important crossover hemoglobin, contains a non–α-globin composed of amino sequences of γ-globin and carboxyl sequences of β-globin (see Fig. 21-18 ). Molecular analysis demonstrated that the A γ-globin gene was involved in the crossover. The crossover occurred approximately at the position of amino acid codon 100. Hb Kenya was first observed in an individual who was heterozygous for a β S gene. This patient had an increased level of HbF (up to 6%) that was uniformly distributed in all his red blood cells. The HbF was entirely of the G γ type. Hb Kenya accounted for 17% to 20% of the total hemoglobin. Persons who are heterozygous for Hb Kenya without HbS are clinically healthy. Approximately 10% Hb Kenya and 6% to 10% HbF of the G γ type are found homogenously distributed in their red blood cells, in contrast to the distribution of HbF with the Lepore-type deletion.
Silent Deletions
Deletions that eliminate a single γ-globin gene or the δ-globin gene are unlikely to result in a phenotype that would be detected in hematologic screening, and thus few of these mutations have been detected and described. A clinically silent deletion of the A γ-globin gene was identified through extensive molecular screening of chromosomes in Melanesians. A 7.2-kb deletion involving the δ-globin gene but not the γ- or β-globin gene was reported in Corfu (δβ) 0 -thalassemia in cis to the G→A substitution at IVS-1 position 5. Homozygotes for the IVS-1 position 5 mutation ordinarily exhibit a mild β+-thalassemia intermedia phenotype with 10% to 20% HbA, but homozygotes for Corfu (δβ) 0 -thalassemia display the clinical phenotype of β 0 -thalassemia intermedia with 100% HbF. This finding has been interpreted as evidence that sequences in the deleted region are important in activating the β-globin gene and potentially in suppressing γ-gene expression. Subsequently, however, the identical deletion was identified in cis to a normal β-globin gene. In this circumstance, the deletion leads to failure of δ-globin gene expression but has no demonstrable effect on expression of the γ- and β-globin genes on the same chromosome. Recent work has suggested that this deletion may increase γ-gene expression in cis to the deletion but that translation of the γ gene may be limiting in certain settings, thus helping explain the discrepancy between carriers of the deletion and homozygotes. The previously discussed mapping studies of HPFH and δβ-thalassemia deletions suggest that the regulatory element upstream of the δ-globin is important for γ-globin silencing, consistent with the idea that the Corfu deletion is removing this critical regulatory element and therefore can result in elevated HbF levels.
β 0 -Thalassemia Deletion Mutations
Several deletions remove the β-globin gene and are associated with typical high-HbA 2 β 0 -thalassemia (see Table 21-4 ). Interestingly, deletions that remove the promoter region are associated with somewhat higher HbF levels than usually seen in β 0 -thalassemia heterozygotes (e.g., see Oner and colleagues ). The HbF is distributed in a heterocellular pattern. In contrast, a 0.6-kb deletion at the 3′ end of the β gene that spares the β-globin promoter, seen in Asian Indians, is not associated with an increased level of HbF in heterozygotes. One explanation for the different phenotypes relates to the possibility that an intact β-gene promoter may interact productively with the LCR and thereby effectively compete with an incompletely silenced γ-globin gene at the adult stage. Removal of the promoter may alleviate competition and favor persistent γ-globin expression.
δβ-Thalassemia
Persons heterozygous for these deletions generally have 5% to 20% HbF composed of both G γ- and A γ-globins and distributed in a heterocellular fashion (see Table 21-4 ). The erythrocytes are typically hypochromic and microcytic. The level of HbA 2 is characteristically low in comparison with the modest elevations observed in most persons with β-thalassemia trait. Homozygotes for these deletions produce only HbF yet exhibit a mild β 0 -thalassemia intermedia phenotype with hemoglobin levels of approximately 10 g/dL. The most common of these deletions is referred to as the Sicilian type. The deletion extends from within the δ-globin gene to just beyond the β-globin gene (see Table 21-4 ). As previously discussed, recent work comparing such deletions with the similar HPFH deletions has provided insight into potential regulatory regions needed for HbF silencing and which are bound by the critical HbF silencing factor, BCL11A.
A γδβ-Thalassemia
The phenotype of these deletions in heterozygotes is identical to that of δβ-thalassemia: hypochromic microcytic red blood cells with low levels of HbA 2 and moderately increased G γ-HbF levels (5% to 15%) with a heterocellular distribution (see Table 21-4 ). Homozygotes have β 0 -thalassemia intermedia with 100% G γ-HbF. The 5′ breakpoints in these instances lie between the two γ genes or within the body of the A γ gene, thereby resulting in silencing of the A γ gene together with the δ and β genes. The 3′ end points of several of these deletions have been precisely mapped. Other deletions are very large and extend well beyond the boundary of the β-gene complex (see Table 21-4 ). One interesting mutation of this type, the Indian form (see Table 21-4 ), has two deletions, one involving the γ gene and the second involving a portion of the δ and β genes and intragenic DNA. The segment of DNA between the two deletions is inverted such that the 5′ ends of the γ and δ genes come to be adjacent but in an inverted orientation.
γδβ-Thalassemia
The diagnosis of γδβ-thalassemia should be considered in newborns with hemolytic disease associated with hypochromic red blood cells. In adults, it is also a cause of normal-HbA 2 , normal-HbF β-thalassemia trait. The syndrome was first identified in a newborn with microcytic, hemolytic anemia. Heterozygous β-thalassemia associated with normal levels of HbA 2 and HbF was identified in the father and many relatives. Decreased γ- and ββ-chain synthesis was demonstrated in the infant’s reticulocytes. The anemia was self-limited; as the baby grew older, the hemolytic anemia disappeared and the phenotype of simple heterozygous β-thalassemia developed in the child.
The syndrome of γδβ-thalassemia results from large deletions within the β-globin gene cluster (see Table 21-4 ). They can be divided into two categories: extremely large deletions that remove the entire β-globin gene cluster or all of the structural genes and deletions such as the Hispanic, English, Dutch, and Anglo-Saxon forms in which the structural genes are left intact but the LCR elements are removed. The remaining genes on the chromosome are not expressed and are found in inactive chromatin that is methylated and resistant to nuclease digestion. Study of these deletions has provided the most convincing evidence that the LCR is required in normal erythroid cells for transcription of the β-like globin genes in vivo. Homozygous γδβ-thalassemia is expected to be incompatible with survival and has not been observed.
β-Thalassemia Mutants Unlinked to the β-Globin Complex
Until recently, the mutations characterized in β-thalassemias resided within or near the β-globin gene itself or the β-globin locus. In principle, defects in trans -acting regulatory factors could lead to failure of proper β-globin gene expression. Two examples are now known.
Mutation of the xeroderma pigmentosum disease (XPD) helicase gene, which encodes a component of the general transcription factor TFIIH, results in β-thalassemia in association with trichothiodystrophy. Patients with specific mutations in XPD have typical features of β-thalassemia trait, including reduced levels of β-globin synthesis. Inadequate expression of diverse, highly expressed genes is presumed to be the direct consequence of an abnormality in TFIIH.
Mild β-thalassemia trait in association with thrombocytopenia was reported as an X-linked trait in a rare family. Linkage studies demonstrated that the gene mutated in this family resides on the short arm of the X chromosome, near the Wiskott-Aldrich syndrome ( WAS ) gene and the erythroid/megakaryocytic transcription factor GATA1. Subsequently, it was shown that affected boys in this family have a specific amino acid substitution in the amino zinc finger of GATA1 that subtly alters its interaction with DNA and leaves its physical interaction with the cofactor FOG-1 intact. How this change in the properties of GATA1 leads to a slight imbalance in globin gene expression is unknown, although it has been suggested that this mutation may disrupt interactions of GATA1 with other erythroid transcription factors such as TAL1. More recently, similar mutations have been found in other patients. In one case, β-thalassemia was present along with an elevation in HbF to nearly 60%. This patient also had features of erythropoietic porphyria and mild thrombocytopenia. However, another patient with a similar mutation had features of only gray platelet syndrome, and β-thalassemia was not present. These phenotypic differences may be due to differential effects on the TAL1 complex to which GATA1 binds.
Hereditary Persistence of Fetal Hemoglobin
Persons who are heterozygous or homozygous for mutations that enhance HbF are asymptomatic. They are identified incidentally in routine screening programs or during investigation of family members with hematologic disease because of interaction of these variants with other mutations in the β-globin gene cluster. Homozygotes for deletion forms of HPFH have normal hemoglobin concentrations, but their red blood cells are slightly hypochromic and microcytic. Minimal globin chain biosynthetic imbalance may be seen. No symptoms or physiologic impairment has been associated with the exclusive production of HbF despite its high oxygen affinity.
Hereditary Persistence of Fetal Hemoglobin Deletion Mutations
Several deletions produce the HPFH phenotype of increased levels of HbF in otherwise normal red blood cells ( Table 21-5 ). * Heterozygotes for these deletions can produce greater than 30% HbF containing both G γ and A γ chains in a pancellular distribution. Differences in the relative percentages of G γ and A γ chains can be detected between different types of HPFH deletions (see Table 21-4 ). Heterozygotes have normocytic red blood cells. The red blood cells of homozygotes contain HbF exclusively. The higher oxygen affinity of HbF may lead to slightly elevated hemoglobin levels in such individuals. Although homozygotes are clinically healthy, the α-globin to γ-globin chain biosynthetic ratio may be slightly imbalanced, and mild microcytosis and hypochromia may be observed. This finding suggests that γ-chain synthesis occurs at levels below the output of the normal β-globin gene on these chromosomes.
Several hypotheses have been put forward to account for the increased expression and pancellular distribution of HbF in the deletion HPFH syndromes, particularly when compared with the moderate increase and heterocellular distribution in δβ-thalassemia. As previously discussed, recent studies have examined the differences between several deletional forms of HPFH and δβ-thalassemia. These studies have led to the identification of a region upstream of the δ-globin gene that appears to be a critical binding site for BCL11A and several partner proteins. This observation has also been substantiated through the analysis of the patients with deletional forms of β-thalassemia, where variation in the level of HbF expressed appears to be due to the presence or absence of this upstream region. Further studies of this sort are needed to better understand the exact mechanisms by which this region acts to silence the γ-globin genes.
* See references .
Nondeletion Hereditary Persistence of Fetal Hemoglobin Mutations
Persons with nondeletion hereditary persistence of HbF mutations exhibit elevated levels of HbF with normal red blood cell indices and are identified through hemoglobin screening programs or by study of families in which a segregating high-HbF allele modulates the severity of sickle cell disease or β-thalassemia. In many instances, single base changes have been discovered within the promoter region of either the G γ- or A γ-globin gene. These mutations are postulated to alter the binding of nuclear regulatory proteins and lead to a more favorable interaction of the promoter with the LCR at the adult stage. Alternatively, these mutations may disrupt the binding of repressor molecules that act to silence the expression of the γ-globin gene. Typically, only increased expression of the γ-globin gene in which the mutation is found occurs.
The level of HbF observed in patients with this syndrome varies greatly and ranges from a few percent to 30% in the −175 T→C substitution found in either the G γ- or A γ-globin gene. Similar to the deletion forms of HPFH, the HbF may be found in either a heterocellular or a pancellular distribution ( Table 21-6 and Fig. 21-19 ). The −158 C→T substitution in the G γ-globin gene is associated with a normal level of HbF in otherwise normal heterozygotes but with a high level of HbF in the presence of erythropoietic stress. For example, in Saudi Arabia, patients with sickle cell anemia often have high HbF levels (25% or greater) and mild disease, whereas their parents with sickle cell trait have normal or only slightly elevated levels. Identification of persons with this mutation (or polymorphism) has been aided by the finding that it is linked to a rare subhaplotype. The −158 HPFH substitution also improves the clinical course of β-thalassemia. A Chinese person homozygous for the −29 promoter mutation has transfusion-dependent β-thalassemia, yet black patients homozygous for the same mutation who co-inherit the −158 HPFH mutation in the G γ promoter have mild β-thalassemia.
| Syndrome | Clinical Features | Hemoglobin Pattern | α-Globin Genes Affected by the Thalassemia Mutation |
|---|---|---|---|
| Silent carrier (α-thal-2) | No anemia, normal red blood cells | 1%-2% Hb Bart’s (γ 4 ) at birth; may have 1%-2% Hb Constant Spring; remainder HbA | 1 |
| Thalassemia trait (α-thal-1) | Mild anemia, hypochromic and microcytic red blood cells | 5%-10% Hb Bart’s (γ 4 ) at birth; may have 1%-2% Hb Constant Spring; remainder HbA | 2 |
| HbH disease | Moderate anemia; fragmented, hypochromic, and microcytic red blood cells; inclusion bodies may be demonstrated | 5%-30% HbH (β 4 ); may have 1%-2% Hb Constant Spring; remainder HbA | 3 |
| Hydrops fetalis | Death in utero caused by severe anemia | Mainly Hb Bart’s; small amounts of HbH and Hb Portland also present | 4 |
Confirmation that the base substitutions in the promoter region are the cause of increased γ-chain synthesis rather than associated random DNA polymorphisms is based on several lines of evidence (reviewed by Ottolenghi and colleagues ). As mentioned earlier, they are generally associated with overexpression of the gene in which the mutation is found and typically represent the only sequence change. None of the mutations listed, with the exception of the −158 mutation, have been observed in persons with normal HbF levels. Haplotype analysis in some pedigrees of patients with HPFH has demonstrated that the HPFH determinant is linked to the β-globin cluster. The British type of nondeletion HPFH is quite informative in this regard. The gene has been tracked through three generations, and three homozygous individuals have been observed. Haplotype analysis using restriction enzyme polymorphisms has established linkage of the British HPFH phenotype to the β-globin gene locus. More than 90% of the γ chains are of the G γ type, and there is an associated mutation at −198 of the G γ gene. Even the three homozygotes, with approximately 20% HbF, have heterogeneous distribution of HbF among their red blood cells. Two homozygotes for −117 A γ HPFH have also been described, and they had 24% HbF in a pancellular pattern. More recent transgenic experiments provide support for the idea that the single base substitutions seen in HPFH can lead to enhanced γ-globin expression in adult life.
An interesting aspect of this syndrome is a balanced α-globin to non–α-globin chain synthetic ratio, even in homozygotes, which implies that increased γ-globin synthesis is offset by decreased β-globin synthesis. Expression of the β gene in cis to the mutation may be reduced by 20% to 30%. It has also been observed that HbA 2 levels are uniformly low in the nondeletion HPFH syndromes and generally correlate inversely with HbF levels. The reduction in δ- and β-globin gene expression in cis to the HPFH mutations is compatible with models of competitive regulation of β-globin expression by the linked γ-globin gene.
Mutations That Alter α-Globin Gene Regulation
In contrast to the β-globin cluster, in which single nucleotide substitutions are the most common cause of thalassemia, large deletions within the α-globin cluster are the predominant basis of α-thalassemia. The overall impairment in α-globin chain synthesis that results from defects in the α-globin cluster is determined by the number of genes inactivated (either by deletion or mutation), the type of lesion (deletion or mutation), and whether the lesion affects the α 2 or α 1 gene.
The α-globin cluster on chromosome 16 contains three functional genes (ζ, α 2 , and α 1 ), an expressed gene of no apparent significance, and three pseudogenes (see Fig. 21-2 ). Transcription of the genes depends on the integrity of the distant regulatory element HS-40. An α-thalassemia deletion mutation (αα RA ) has been reported that removes a large segment of DNA upstream of the ζ 2 gene but spares the remainder of the cluster. In heterozygotes, no expression of the ζ- or α-globin genes from this chromosome is detected in heterozygotes. These findings are formally analogous to those in Hispanic and English γδβ-thalassemias (see Table 21-4 ), in which upstream LCR elements of the β-globin cluster are deleted.
The two human α-globin genes are thought to have been generated through a gene duplication event that occurred approximately 60 million years ago. The nucleotide sequences of the α 2 and α 1 genes have remained remarkably similar and represent an example of concerted evolution in which the two genes have exchanged genetic information through crossover fixation and gene conversion events. The coding regions of the two genes are virtually the same and encode identical polypeptides. The genes differ only in minor respects within IVS-2, whereas the sequences diverge significantly in the 3′-untranslated region.
The α-globin genes are expressed in embryonic, fetal, and adult-stage erythroid cells. Initially it was believed that the two α genes were expressed at similar levels, but subsequent RNA analysis relying on sequence differences in their 3′-untranslated regions revealed that α 2 RNA predominates over α 1 RNA by a ratio of 3 : 1. Because the two α-globin mRNA molecules have the same intrinsic stability, the higher level of α 2 RNA reflects increased transcription of the α 2 gene. “Transcriptional interference” of the α 1 gene by the upstream α 2 gene has been proposed as an explanation for this observation. Ribosome-loading studies suggest that α 2 and α 1 transcripts are translated at equivalent rates. Systematic characterization of the expression of α-globin structural variants at both the α 2 and α 1 loci has confirmed that the α 2 gene has a predominant role in α-globin chain production. A direct prediction of this model is that mutations altering the α 2 gene would result in a greater deficiency of α-globin chain production than would mutations of the α 1 gene. Clinical support for this prediction is provided by study of Sardinian patients with HbH disease who are heterozygous for one chromosome with a deletion of both α genes (−/) and a chromosome with a nondeletion mutation affecting the Inr codon of either the α 2 gene (α T α/) or the α 1 gene (αα T /). Patients with the mutation in the α 2 gene (−/α T α) have disease that is clinically more severe. Consistent with its predominance, the α 2 gene is involved in the majority of the reported mutations of the α-globin genes (see Table 21-2 ).
Deletion Mutations within the α-Globin Gene Cluster
Mutations That Remove One α-Globin Gene
The two α-globin genes are embedded in highly homologous, tandem repeated sequence blocks (called X, Y, and Z) that are separated by nonhomologous segments ( Fig. 21-20 ). Unequal, homologous recombination through the X and Z blocks generates a chromosome with a single α-globin gene and another with three α-globin genes. Homologous recombination in the small Y box has not been observed. The most common type of deletion in this class removes 3.7 kb as a result of misalignment of the Z boxes and is known as “rightward deletion.” The products of this crossover are the (−α 3.7 /) and (ααα anti-3.7 /) haplotypes. The −α 3.7 products may be further subdivided into types I, II, and III by the precise location of recombination within the Z box. Unequal crossover events through the X box leads to “leftward deletion” of 4.2 kb of DNA and the −α 4.2 chromosome and its triplicated antitype. The incidence of the observed recombination products appears to reflect the size of the homologous target sequence within the boxes because −α 3.7I (1436 bp) is the most common, followed by −α 4.2 (1339 bp), −α 3.7II (171 bp), and −α 3.7III (46 bp). Unequal α-gene recombination through the X and Z boxes has been reproduced in both prokaryotic and eukaryotic experimental systems with episomal vectors.