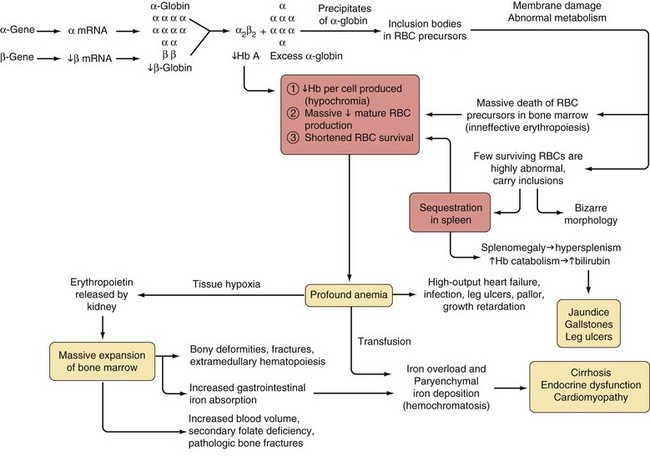
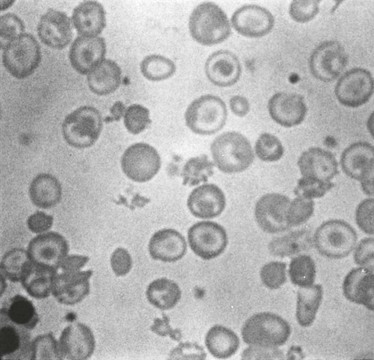
Chapter 13 Thalassemia Syndromes
Table 13-1 Common β-Thalassemia Mutations in Different Racial Groups
| Racial Group | Description |
|---|---|
| Mediterranean | IVS-1, position 110 (G → A) |
| Codon 39, nonsense (CAG → TAG) | |
| IVS-1, position 1 (G → A) | |
| IVS-2, position 745 (C → G) | |
| IVS-1, position 6 (T → C) | |
| IVS-2, position 1 (G → A) | |
| Black | -34 (A → G) |
| -88, (C → T) | |
| Poly(A), (AATAAA → AACAAA) | |
| Southeast Asian | Codons 41/42, frameshift (-CTTT) |
| IVS-2, position 654 (C → T) | |
| -28 (A → T) | |
| Asian Indian | IVS-1, position 5 (G → C) |
| 619-bp deletion | |
| Codons 8/9, frameshift (++G) | |
| Codons 41/42, frameshift (–CTTT) | |
| IVS-1, position 1 (G → T) |
Data from Kazazian HH Jr, Boehm CD: Molecular basis and prenatal diagnosis of beta-thalassemia. Blood 72(4):1107, 1988; and Kazazian HH Jr, Boehm CD: personal communication, 1993.