Chapter Outline
Conventional Chemotherapeutic Approaches: Not Entirely Nonspecific
Lessons Learned from Cytotoxic Chemotherapy
INTRODUCTION TO TARGETED THERAPY
NEW AND EMERGING OPPORTUNITIES IN PEDIATRIC CANCER
Special Considerations in Targeted Therapy Development for Children
Emerging Genetic Targets in Pediatric Cancer
Emerging Lineage-Dependent Targets in Pediatric Cancer
Emerging Synthetic Lethal or Empiric Track: Targets in Pediatric Cancer
With the discovery of nitrogen mustard as an anticancer agent in the 1940s, medicinal and hence systemic cancer therapy became a reality. The identification and application of cytotoxic agents dominated the next 60 years of cancer treatment. The success of imatinib for BCR-ABL rearranged chronic myelogenous leukemia (CML), the dramatic responses seen in patients with BRAF -mutated melanoma and ALK rearranged non–small cell lung cancer to targeted inhibitors, and the unparalleled ability to systematically characterize cancer genomes have catapulted the community into a new era of targeted cancer therapy. Nonetheless, there is much to learn from the 60-year experience in empiric-based therapy that should be directly applicable to the development of molecularly targeted therapy. Cytotoxic chemotherapy, although often regarded as largely unsuccessful, has led to substantial cure rates in a number of well-defined malignancies, particularly pediatric. In this chapter we will discuss the fundamental concepts, advantages, and disadvantages of cytotoxic cancer therapy and their relevance to targeted drug therapy for malignancy. We will define targeted therapy, its advantages and disadvantages, discuss the credentialing of targets, and provide a system for categorizing classes of targets and classes of therapies. We will then focus on particular lessons learned in the early development and testing of new targeted therapies for adult malignancies, with examples from each of the target classes. Our intent is to highlight illustrative examples rather than to provide an exhaustive list of all targeted therapies in development. We will conclude with a discussion of the challenges facing us in the development of targeted therapies for children and highlight emerging pediatric cancer-based targets.
Traditional Chemotherapy
Conventional Chemotherapy: Not Entirely Nonspecific
Although conventional chemotherapy agents are often seen as nonspecific cytotoxic drugs, over the years more information has come to light about their mechanisms of action in killing tumor cells. In general, these agents were designed to interfere with molecular pathways required for nucleotide synthesis, DNA replication, and the mechanics of cell division. Thus these agents have significant but often predictable, or at least stereotypical, morbidity related to target-mediated suppression of hematopoiesis or of the cell-division process required for homeostasis of other organ systems. These agents are discussed in greater detail in Chapter 45 .
Lessons Learned from Cytotoxic Chemotherapy
The use of cytotoxic chemotherapy agents is an imperfect science at best, but researchers have learned many lessons over the years about the medicinal treatment of cancer, lessons to strongly consider in the development of targeted therapy ( Box 44-1 ).
- 1.
Combination therapy is required for cure.
- 2.
Appropriate dose intensity is required for improved outcome.
- 3.
Toxicity is acceptable when therapy is curative.
- 4.
Establish curative regimens, and then attempt to reduce toxicity.
Combination Therapy Is Required for Cure
The goals of combination therapy are numerous. First, because it is difficult to predict which single agent will be most effective against an individual patient’s tumor, the use of multiple drugs increases the potential for an initial complete response. Second, combination therapy is likely critical for preventing the development of therapeutic resistance. Third, drug combinations may have additive or synergistic interactions, increasing the total activity of the drugs and potentially enabling lower dosing and reduced toxicity. One example is the successful dose reduction of cytotoxic chemotherapy with the addition of all- trans -retinoic acid (ATRA) for the treatment of patients with acute promyelocytic leukemia (APL). With few exceptions, curative systemic therapy has required the combination of highly active agents rather than combinations of active and inactive agents.
The need for multiagent therapy was first appreciated in the treatment of acute lymphoblastic leukemia (ALL). At best, 60% of patients are estimated to achieve complete remission with single-agent chemotherapy. Despite continued use, almost all patients will relapse within 6 to 9 months with single-agent therapy. However, complete remission rates exceed 95% in patients treated with combination chemotherapy, and cure rates today for children with ALL exceed 80% with multiagent treatment regimens. Similarly, patients with APL had very poor remission and cure rates until the discovery of ATRA differentiation therapy for this disease. ATRA achieved a high complete remission rate in use as a single agent, but virtually all patients ultimately relapsed. It was not until the combination of cytotoxic chemotherapy with ATRA and the recent combination of ATRA with arsenic trioxide that these patients obtained the best long-term survival rates of any acute myeloblastic leukemia (AML) subtype.
Appropriate Dose Intensity Is Required for Improved Outcome
A guiding principle in the delivery of cytotoxic chemotherapeutic agents is the concept of the maximum tolerated dose (MTD): the simple idea that more is better. Drugs are delivered at the maximum dose possible, a dose greater than which unacceptable toxicity is encountered. Here, without a clear cellular biomarker of cytotoxic activity, it is not possible to relate the maximum molecular effect to maximum achievable efficacy. Thus cytotoxic drugs are first evaluated in phase I studies with the primary study goal of establishing the MTD. Subsequent phase II and III trials are then based on this dosing schedule. MTD assumes that some toxicity is acceptable and, in fact, expected in the pursuit of cure. The most extreme example of this concept has been the use of autologous stem cell transplantation to deliver what otherwise would be lethal doses of chemotherapy. In this case the MTD is surpassed by replacing the dose-limiting factor, the hematopoietic system, with stem cells harvested from the patient before delivery of the conditioning chemotherapy. While clearly a brute force approach, the MTD paradigm also established an important corollary: significant side effects can be tolerated when therapy is administered with curative intent (discussed later).
Many retrospective studies of chemotherapy dose intensity and outcome support this premise, particularly in the pediatric setting. In high-stage neuroblastoma the addition of dose intensification with stem cell transplantation has improved the outcome for patients with metastatic disease. In pediatric ALL several studies suggest that dose intensification is important for long-term outcome, including one randomized study comparing standard versus dose-reduced methotrexate with mercaptopurine. Moreover, in Ewing sarcoma chemotherapy intensification through interval compression in a randomized trial was also associated with an improved outcome in patients with localized disease. However, fewer prospective studies than retrospective studies have been performed to address the issue of dose intensity and outcome for pediatric tumors. Significantly, while these studies begin to emerge, some previous assumptions about MTD have come into question. For example, retrospective studies support the notion that doxorubicin or methotrexate dose intensity is important for outcome in patients with localized high-grade osteosarcoma. However, prospective studies involving large patient numbers have called into question the role of further dose intensification for this disease. Furthermore, randomized trials for malignancies such as breast cancer have demonstrated that massively increasing dose with stem cell transplantion does not always improve long-term survival. Well-controlled prospective trials are critical to address the issue of dose intensification, even for cytotoxic agents.
Toxicity Is Acceptable When Therapy Is Curative
The oncology community has accepted toxicity as inherent in the mission of cure. With the implementation of dose-intensive regimens, however, comes additional toxicity. Because of the relatively nonspecific nature of traditional chemotherapy, morbidity from these treatment regimens is significant. Acute toxicity takes the form of infection, transfusion dependence, renal injury, and hepatic injury. There are also potential long-term consequences such as cardiac injury, infertility, chronic renal insufficiency, hearing loss, skeletal injury, and secondary malignancies. Ongoing efforts are focused on decreasing these short- and long-term side effects. For example, to minimize acute toxicity, chemotherapy dosing is usually individualized on the basis of body surface area (mg/m 2 ). For some cytotoxic agents, such as methotrexate and busulfan, drug levels can be monitored to minimize toxicity. Dose intensity can also be achieved with regional tumor delivery, growth factor support, or rescue with stem cells. Furthermore, better supportive care, such as infection prophylaxis, has enabled tolerance of prolonged immunosuppression. To decrease long-term morbidity, “safe” maximum cumulative doses are being established, such as maximum cumulative doxorubicin to minimize late cardiac effects. Although toxicity can be tolerated, it is essential that the level of toxicity be commensurate with the level of benefit. Marginally effective therapeutic regimens that result in substantial toxicity should not become accepted standards.
Establish Curative Regimens and Attempt To Reduce Toxicity
Treatment strategies for most of the pediatric malignancies, including ALL and the solid tumors, have used disease stratification to decrease dose in treatment regimens. A dose-intensive regimen is initially used to establish efficacy. Then, patients with a good prognosis are treated with less intensive regimens, with the goal of comparable disease-free outcomes but decreased treatment-related morbidity. This is particularly important for pediatric disease, in which the long-term toxicity of treatment is experienced over a lifetime. Historically, these stratifications were based on factors such as age, stage, and histology, but now they include molecular genetic markers, such as MYCN amplification in neuroblastoma, and MLL rearrangement in pediatric ALL. For example, the National Wilms’Tumor Study evaluated a half-dose chemotherapy regimen for infants with low-risk disease in an attempt to decrease the incidence of toxic deaths without compromising long-term survival. These infants had acceptable toxicity with no toxic deaths, and long-term survival was not compromised. A similar approach has been taken with infants and young children with low-stage neuroblastoma, and therapy for children with ALL has been dose-reduced based on rapid clearance of minimal residual disease.
While our understanding of the molecular basis of malignancy improves, so will our ability to develop fine-grained prognostic categories and patient-tailored treatment regimens, perhaps even for traditional chemotherapy agents. While we implement targeted therapy, it is expected that disease stratification and patient selection will be based on molecular genetic findings related to the credentialed target. In the clinical development of targeted therapies, we must continue to consider these key concepts: combination therapy, appropriate dose intensification, tolerance of toxicity for substantial efficacy and cure, and treatment adjustment within established favorable prognostic groups.
Shortcomings of Cytotoxic Chemotherapy
Although clinicians have made great progress with the cytotoxic armamentarium, further strides in the treatment of cancer are limited with this approach alone. Several shortcomings of this approach are intimately related to the lack of specific target knowledge ( Box 44-2 ).
- 1.
Toxicity is significant.
- 2.
Optimization of anticancer effect versus side effects is difficult.
- 3.
Mechanistic understanding of nonresponding and resistant disease is lacking.
- 4.
Predictors of patient response to a specific drug are poor.
- 5.
To date cytotoxics have explored a narrow range of cellular and molecular mechanisms.
- 1.
Toxicity is significant.
- 2.
Optimization of anticancer effect versus side effects is difficult.
- 3.
Mechanistic understanding of nonresponding and resistant disease is lacking.
- 4.
Predictors of patient response to a specific drug are poor.
- 5.
Cytotoxics have explored a narrow range of cellular and molecular mechanisms.
One major shortcoming to cytotoxic therapy is morbidity. Because these agents are relatively nonspecific, there is significant toxicity to normal host cells. Patients can expect to experience short- and long-term morbidity from these drugs and even incur the risk of therapy-related fatality. Furthermore, because the most relevant targets of these drugs are poorly understood, it is difficult to optimize the anticancer effect versus the toxicity, and it is difficult to separate on-target from off-target toxicities. Often, there is no relationship between the administered dose (pharmacokinetics) or the MTD and inhibition of the putative target (pharmacodynamics). Developing a strong correlation between the pharmacokinetics of a drug and its pharmacodynamic effect is obviously impossible when the target of a drug is unknown. Similarly, in the absence of target knowledge, our understanding of resistant or refractory disease is poor at best, and it is very difficult to predict responders a priori. This has necessitated long clinical development programs with large trials to identify responsive subpopulations of patients and then follow-up trials to more thoroughly investigate response in this patient subset. Targeted therapy has the potential to address these shortcomings, to enable more rational tumor-specific therapy with less morbidity risk, and to explore previously unexploited cancer-related molecular mechanisms. With the sequencing of the human genome, the development of high-throughput genomic technologies, and an ever-increasing understanding of the molecular pathogenesis of cancer, this promise is likely to be realized.
Introduction to Targeted Therapy
What Is Targeted Therapy?
Although we have made great strides with the cytotoxic armamentarium, further strides in the treatment of cancer are limited with this approach alone. We still fail to cure many patients with this approach, and several shortcomings of this approach are closely related to the lack of specific target knowledge (see Box 44-2 ). Targeted therapy, defined as a therapeutic procedure whose preclinical development focused on modulation of a single molecular target (typically inhibition but occasionally activation of the molecular entity), has the potential to address these shortcomings. Targeted therapy relies on the concept of cancer cell dependence—that is, the malignant cell or tumor as a whole depends for its survival or proliferation on the particular gene product or pathway. The discovery of targeted therapy differs significantly from that of cytotoxics. Historically, cytotoxic therapy discovery relied on simple, phenotypic cell death assays in which small-molecule libraries were screened in vitro against a panel of cancer cell lines. Because cytotoxic compounds are believed to cause nonspecific cell death, effective compounds should, in principle, have efficacy for all cancer types. In practice, however, this has not proved to be the case. Thus although phenotypic screens may be of significant value in identifying molecules with complex mechanisms of action, the direct prediction of clinical efficacy from limited in vitro testing remains problematic. This is in sharp contrast to the concept of targeted therapy.
There are three critical elements to the implementation of targeted therapy. First, on the basis of available data, a hypothesis is generated suggesting a causal or pathogenic link between a specific protein or gene (the potential drug target) and cancer. Experiments are then performed and data generated to test the validity of this hypothesis. Is this candidate target relevant to oncogenesis or the maintenance of the malignant phenotype? Second, there must be a valid therapeutic approach to interfering with the target activity through small molecules, antibodies, or protein- or cell-based biologics. Third, the clinician must estimate the likely impact of target manipulation in a relevant clinical application. For example, a targeted therapeutic may have limited clinical application if the disease state is already readily cured. The process of evaluating potential targets that emerge is target “validation.”
Advantages of Targeted Therapy
Targeted therapy offers several potential advantages over nonspecific cytotoxic treatments ( Box 44-3 ).
- 1.
Improved patient selection is enabled.
- 2.
Drug efficacy and target inhibition are linked, enabling rational dosing.
- 3.
Side effects are more predictable.
- 4.
Understanding of target and pathway increases options for the subsequent development of therapeutics with improved efficacy.
- 5.
The mechanisms of resistance may, in some cases, be more readily discovered and interdicted with second-generation therapeutics or specific combinations.
- 6.
Rational pathway-based combination regimens can be tested.
- 1.
Improved patient selection is enabled.
- 2.
Drug efficacy and target inhibition are linked, enabling rational dosing.
- 3.
Side effects are more predictable.
- 4.
Understanding of target and pathway increase options for the subsequent development of therapeutics with improved efficacy.
- 5.
The mechanisms of resistance may, in some cases, be more readily discovered and interdicted with second-generation therapeutics or specific combinations.
- 6.
Rational pathway-based combination regimens can be tested.
By way of example, hormonal therapy has become standard of care for patients with estrogen receptor (ER)–positive breast cancer. In this case there is a lineage-dependent target in the malignant cell, the ER, and antiestrogen agents, such as tamoxifen, have been developed to inhibit competitive binding of estrogen to the ER. Numerous studies exploring estrogen antagonist therapy with tamoxifen for patients with ER-positive versus ER-negative disease demonstrate benefit only in the ER-positive patients. In conjunction with diagnostic tools to identify hormonal receptor status, patients are now rationally chosen for therapy, thus eliminating exposure to drug toxicity for patients not likely to respond.
A second advantage of targeted therapy is the strong link between the target inhibition and efficacy. The dosing goal is achievement of complete target inhibition, not MTD. Thus there is no need to suffer undue toxicity in the achievement of an MTD. In fact, studies have shown that tamoxifen treatment responses do not correlate with serum levels and that doses greater than 20 mg daily do not increase efficacy compared with that of this dose. A related third advantage of targeted therapy is that the side effects can generally be anticipated and serve as a marker for on-target inhibition. For example, the side effects of tamoxifen are predominantly related to the antiestrogen effects of this drug: vaginal dryness, hot flashes, and irregular menses. Because this drug also has proestrogen effects, thromboses and endometrial cancer can be seen.
A fourth advantage of targeted therapy is that understanding the target and the pathway increases the possibilities for therapeutic design. In the case of modifying ER signaling, there are several methods to accomplish this other than direct ER antagonism. One method is to decrease ovarian production of estrogen with luteinizing hormone–releasing factor agonists, which induce a medical ovarian ablation. A second method of interfering with ER signaling is to decrease estrogen production through the inhibition of aromatase activity. Aromatase is the final enzyme in estrogen synthesis converting the androgens androstenedione and testosterone to the estrogens estrone and estradiol. Aromatase inhibitors work by decreasing this conversion and thus decreasing circulating estrogens. Aromatase inhibitors have improved efficacy over tamoxifen and have the advantage of fewer side effects in terms of hot flashes, endometrial disease, and ischemic cerebrovascular effects. Because of the clear dependence of some breast cancers on ER and detailed knowledge regarding estrogen biosynthesis and signaling, multiple agents could be developed to inhibit this pathway.
The fifth advantage of targeted therapeutics is the ability to more readily understand the molecular mechanisms associated with resistance. The success of targeted inhibition of BCR-ABL in CML and of epidermal growth factor receptor (EGFR) in lung cancer was rapidly followed by the specific elucidation of direct resistance mutations in the target leading to decreased activity of the relevant small-molecule inhibitors, imatinib and erlotinib or gefitinib, respectively. In the case of CML, new agents were developed in short order to treat resistance in patients by more efficient inhibition of the mutant BCR-ABL proteins.
The final advantage of targeted therapies may be the ability to generate rational and novel combination regimens. Currently, the vast majority of combination strategies derive from combining a novel agent with components of existing standards of care without regard to mechanism. This strategy is largely based on clinical expedience, existing clinical efficacy (for the standard of care) and tolerability. The difficulty of empirically discovering new synergistic combinations stems from the large number of permutations required to test all possible combinations against multiple in vitro and in vivo models or multiple cancer types. Targeted therapeutics are often related to one another in the context of a pathway. This allows testing of specific pathway-related hypotheses. For example, combinations might target upstream and downstream components of the same pathway to defeat feedback mechanisms, target components of parallel pathways to defeat pathway redundancy, or target distinct antiproliferative and prosurvival pathways.
Classes of Targets
Much of current cancer therapy development focuses on specific changes that alter the malignant cell, setting it apart from its normal counterpart and creating distinct molecular or pathway dependence for cell survival and replication. These may be genetic mutations, gene rearrangements, epigenetic modifications such as altered patterns of methylation or acetylation, lineage legacies, or other metabolic liabilities. Genes whose expression and activation have been increased, resulting in a cancer-dependent state, are prime targets for therapy development. On the basis of cancer dependencies, cancer targets can be categorized in four classes: genetic, lineage, host, and synthetic lethal or empiric ( Box 44-4 ). Although some targets may cross over from one class to another, this classification may be useful in the design of clinical trials and elucidation of the parallel biomarker strategy.
- 1.
Genetics track
- 2.
Lineage track
- 3.
Host track
- 4.
Synthetic lethal or empiric track
Genetics Track
The genetics track relies on the idea that a cancer develops from a finite number of genetic alterations, that these alterations give the malignant cell a selective advantage, and that these genetic changes are selected for during malignant cell replication. Hence the malignant cell becomes dependent on a set of these changes for continued survival, a process sometimes referred to as oncogene addiction. This cancer cell dependence or addiction should provide a window for drug specificity for the malignant cell compared with normal cells. Targeted therapeutics taking advantage of genetic abnormalities should, in theory, be more injurious to the malignant cell than to the normal cellular counterpart. Advances in cytogenetic studies, high-throughput gene sequencing, detection of copy number alteration, whole genome expression profiling, and proteomic approaches have revolutionized candidate genetic target discovery. Now, tens of thousands of genes in hundreds of tumor samples can be analyzed simultaneously. These candidate targets must then be carefully credentialed for their suitability as a drug target. Is there cancer cell dependence on the genetic abnormality, and can the target be pharmacologically manipulated? Imatinib is one of a number of examples of clinically validated targeted therapeutics exploiting a genetic dependence, in this case the dependence of CML on the activity of the Abelson kinase (ABL). The very existence of the cancer-dependent state is dramatically illustrated by the emergence of resistance mechanisms that largely appear to restore Abelson activity (see further discussion later in this chapter).
Lineage Track
Cancer cells often maintain developmental features of the lineage from which they were derived. This is well supported with gene expression studies that report tumor cells more closely resembling the normal lineage counterpart than tumors of a different cell type. The notion that lineage or “legacy” features of a tumor cell may be exploited with targeted therapy is known as lineage addiction . One of the earliest examples of the success of this approach has been the antagonism of ER and its signaling pathway in patients with ER-positive breast cancer. Similarly, the vast majority of patients with prostate cancer derive significant benefit from antagonism of the androgen receptor (AR). In both ER-positive breast cancer and in prostate cancer (virtually all forms of which express AR), the emergence of hormone-refractory disease largely appears to rely on mechanisms that restore steroid receptor signaling. Thus as in the case of imatinib resistance, the dependent state is illustrated by these resistance mechanisms. In many cases lineage dependence may also derive from the normal hierarchy of differentiation beginning with the stem cell.
Host Track
The importance of tumor cell environment to the development and maintenance of the malignant state has become increasingly recognized and represents another potential inroad to targeted therapy. It is possible that tumors, by evolving in a particular environmental context, may become dependent on certain growth factors or neighboring cells for survival in that niche. Preclinical evidence suggests that many tumors require de novo blood vessel development for their growth and maintenance. The identification of these angiogenesis factors has enabled the development of modifiers of the angiogenesis process, leading to the successful clinical application of bevacizumab, an anti–vascular endothelial growth factor (VEGF) antibody, in patients with colorectal cancer and the approval of kinase insert dependent receptor inhibitors (sunitinib and sorafenib) in renal cell carcinoma (see further discussion later in this chapter). The interaction of tumors with the bone microenvironment has also been targeted using inhibitors of osteoclastogenesis. Here, multiple bisphosphonate agents are approved for the prevention of cancer-related fractures.
Synthetic Lethal or Empiric Track
A number of highly efficacious anticancer agents do not conveniently map to the first three categories. Admittedly, we can say that the understanding of the mechanisms of efficacy of such agents remains opaque. Nonetheless, it is quite likely that such agents, including many of the cytotoxic agents described previously, take advantage of aspects of cancer cell biology that remain unknown but may be broadly termed as synthetic lethal interactions. Synthetic lethal genetic interactions are defined as mutations that are lethal in combination but that do not produce a lethal phenotype as a single event. In a similar way, gain-of-fitness alterations in a malignant cell, which give the cell a survival advantage, can sensitize the cell to other stresses that have no consequence in a normal state but may be lethal with the malignancy promoting alteration. Thus cancer-promoting mutations give the malignant cell a survival advantage, but they may also incur a distinct liability that can be exploited therapeutically. Rather than two synthetic lethal genetic events, the clinician can also apply such synthetic lethal concepts to a preceding cancer-related molecular alteration followed by the application of a therapeutic. For example, a striking synthetic lethal relationship was discovered between loss of BRCA1 or BRCA2 function and inhibition of the poly(ADP-ribose) polymerase I enzyme (PARP). Here, loss of PARP1 was found to induce double-strand DNA breaks that required the recruitment of the RAD51 homologous recombination repair complex to the site of these breaks for effective repair. Because it was known that BRCA1 and 2 were required for RAD51 recruitment to such complexes, it was hypothesized that loss of BRCA1 or BRCA2 would create sensitivity to PARP inhibition. This was demonstrated both with siRNA and small-molecule inhibitors of PARP. Moreover, clinical activity of PARP inhibitors has now been seen in patients with BRCA1 and BRCA2 mutations with ovarian or breast cancer.
Credentialing Targets
By the strictest of measures, a drug target is not validated until a selective inhibitor of the target of interest has proven clinical activity and when resistance emerges through specific alterations in the direct target. Although very few cancer targets have achieved this degree of validation, we can envision preclinical experimental methods that will allow the researcher to credential a target for further drug discovery efforts. In this effort, the goals are to build data sets that either prove or disprove certain target-based hypotheses and to use such data sets to prioritize target selection for future drug discovery efforts. We can consider three key questions regarding the relevance of any given target to anticancer therapy:
- 1.
Is the activity of the target required for the development and maintenance of the cancer-dependent state?
- 2.
Is it possible to identify a strategy for developing a therapeutic for the target of interest?
- 3.
Is there a clinical application for the eventual therapeutic?
In the subsequent sections we will focus on the approaches relevant to addressing the role of a given target in cancer dependence. In this specific regard there are two general sources of information: the study of a target in human cancers (target epidemiology) and the functional study of a target in model systems of tumor biology (functional validation) ( Box 44-5 ).
Target epidemiology: study of a target in human cancers
Target expression
Genetic alteration
- •
Gain-of-function
- •
Translocation
- •
Activating point mutations
- •
Copy number gain
- •
- •
Loss-of-function
- •
Translocation
- •
Inactivating point mutations
- •
Deletion
- a.
Homozygous
- b.
Heterozygous
- c.
Loss of heterozygosity
- a.
- •
- •
Protein Epidemiology
Functional validation: study of a target in a model system of tumor biology
Gain-of-function: address sufficiency of target
Loss-of-function: address necessity of target
Target Epidemiology
Preclinical models of cancer are limited in both number and in their qualitative relationship to human cancers. Comprehensive collection of human tumor specimens could, in theory, be used to study a given target in a large array of samples broadly covering tissue distribution and stage distribution (primary versus metastatic sites). In practice, however, there are limited sets of comprehensive tumor collections, and generally the largest tumor-type specific collections are limited to fixed, paraffin-embedded specimens. The availability of tissue samples paired with robust clinical data remains limited. Furthermore, the vast majority of human cancer collections come from surgical resection samples, which do not necessarily reflect the disease state in which a therapeutic will be initially tested. Despite these limitations, molecular epidemiology studies in tissue samples, enabled by tissue microarrays, single nucleotide polymorphism (SNP) arrays, expression microarrays, reverse protein arrays, and next-generation sequencing technologies remain critical to building a case for the cancer relevance of a given target.
Target Expression.
The desire to discover genes “selectively” expressed in cancer led first to the development of differential expression methods, including the subtractive hybridization cDNA libraries, differential display, and serial analysis of gene expression (SAGE) as examples. Following the elucidation of the human genome sequence and the development of DNA-based microarrays, comparative expression profiling largely replaced difference methods, and more recently, RNA sequencing has become a commonly used tool. In all cases the basic premise has been to identify genes overexpressed in cancer with the assumption that overexpression will equate with increased sensitivity to target inhibition and hence serve to improve the therapeutic index. This basic idea, however, must be challenged and carefully considered for each target, for the simple reason that resistance to therapeutics can likewise be engendered through overexpression. Indeed, on the basis of standard biochemical principles, increased target expression should necessitate a higher drug concentration to achieve target inhibition. This is clearly the case in the amplification of dihydrofolate reductase (DHFR) in generating resistance to methotrexate. Likewise, data suggest that increased expression of AR can lead to resistance to AR blockade. Thus caution is required before proceeding with target selection based solely on expression data in the absence of a demonstrated causal or functional link to the maintenance of a cancer state.
Genetic Alterations.
As illustrated by the success of ATRA, trastuzumab, and imatinib, direct genetic alteration leading to gene activation of a given drug target remains perhaps the best evidence for the causal role of a given gene in carcinogenesis. Genetic alterations that are highly frequent in a given tumor type and are present in the majority of cells are a strong indicator of causality. In the era of large-scale resequencing projects, it is critical that one distinguishes “driver” mutations (causal mutations) from so-called passenger mutations. In addition, driver mutations that arise late in the course of disease, are present in only a minor fraction of the tumors, or are present in only a fraction of tumor cells in a given single tumor must be given less weight. These findings would suggest a reduced likelihood of this lesion possessing a strong causal link to the initiation of the disease state, although they may pose an eventual mechanism of drug resistance. The types of genetic alterations (e.g., point mutations, focal amplifications, broad amplifications, and translocations) are also distinguished with respect to the extent to which they can be used to establish causality or to infer dependence.
Gain-of-Function Genetic Alterations.
Gain-of-function genetic alterations include translocations, activating point mutations, and increases in gene copy number. Translocations were among the first discovered genetic alterations initially described using chromosome spreads. Approaches to the discovery of chromosomal rearrangements include exon microarrays, which can potentially detect imbalanced or amplified translocations, cancer outlier profile analysis (COPA), paired-end resequencing of BAC clones from a genomic library (giving a structural map of a cancer genome), and newer methods of single-molecule sequencing (discussed later). Translocations are highly informative with respect to causality because they typically alter only two genes, and because background or random translocation events are relatively rare, with chromothripsis (massive DNA rearrangements affecting one or a few chromosomes that occur as a result of one catastrophic event) as a notable exception.
Amplifications are now routinely detected by methods of DNA copy number analyses: comparative genomic hybridization (CGH) array, high-density SNP arrays, and whole exome or whole genome sequencing. There are two general types of increases in gene copy: focal amplifications and broad copy number increases covering an entire chromosome or chromosome arm. With sufficient sample numbers it is possible to discern one to several genes contained within a focal amplicon. Typically, the minimal region of amplification is a guide. However, methods for detecting copy number alterations (e.g., genomic identification of significant targets in cancer [GISTIC]) also use the amplitude of amplification as an important parameter. Although it is not definitive proof, amplification lends evidence to a causal relationship between a given gene within an amplicon and a cancer state. In some amplicons, multiple genes remain strong candidates for oncogenes targeted by the amplification. One notable example is the 11q13 amplification with genes including Cyclin D1 , FGF3 , and FGF4 .
Larger-scale chromosomal alterations leading to trisomy or gain of an entire chromosome arm are frequent yet are far more difficult to study. As such, identifying single genes (or drug targets) within such broad regions remains a challenge. In this case the number of genes implicated in such large-scale alterations is so large that almost no importance can be ascribed to the cancer relevance of a gene contained within such broad chromosomal-level alterations. Outlier analysis may begin to allow the elucidation of multigene targeting by such broad regions and is illustrated by the analysis of trisomy 7 in glioma revealing a dependence on cMET and its ligand HGF, both located on chromosome 7.
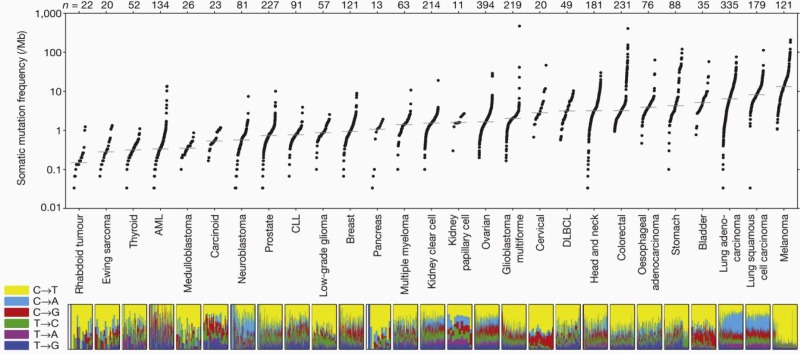
Subtle nucleic acid level alterations, including point mutations and small insertions and deletions, are an important mechanism for the activation of oncogenes. Point mutations leading to the constitutive guanosine triphosphate (GTP)–bound and hence activated form of N-RAS and K-RAS, as well as genetic events leading to activation of the BRAF kinase in melanoma, thyroid cancer, colorectal cancer, pediatric pilocytic astrocytoma, and Langerhans cell histiocytosis are exemplars of this mechanism. Large-scale sequencing projects that unveiled new mutations activating oncogenes such as EGFR and PIK3CA have led to national and international cancer genome sequencing projects. From these studies it has become apparent that the detection of passenger mutations, constituting the background rate of mutation, can pose a significant challenge. These passenger mutations must be separated from the more prevalent and recurrent driver mutations. Such passenger mutations may be exceedingly high in certain types of tumors, particularly those bearing mutations in mismatch repair genes. Indeed, in one case inactivating mutations in the mismatch repair gene MSH6 were discovered in glioma tumors recurring after treatment with temozolomide. Here the sequencing of MSH6 was triggered by the very high rate of observed passenger mutations in treated versus untreated tumors. These data suggest that cautious interpretation of low-frequency events is warranted when considering mutation analysis from larger-scale modalities. A framework for context-specific mutation analysis has been provided by a systematic study of mutation data across many cancer types in The Cancer Genome Atlas (TCGA) project and others, and these studies are revealing remarkably simple genomes in many pediatric cancers ( Fig. 44-1 ).
Loss-of-Function Genetic Alterations.
Loss-of-function alterations do not typically directly indicate or point to a specific gene as a candidate drug target. However, such alterations may predict sensitivity to agents acting against protein targets contained within downstream or parallel pathways. For example, clinical trials are under way testing inhibitors of mammalian target of rapamycin (mTOR) function against sporadic tumors or hereditable cancer predisposition syndromes resulting from loss of the tumor suppressors PTEN and TSC1 or TSC2. In both cases loss-of-function mutations in these tumor-suppressor genes are thought to lead to constitutive activation and hence constitutive dependence on mTOR (discussed later in this chapter).
Similarly, in the hedgehog pathway, a key development pathway regulating cell patterning, deletions or mutations in the gene encoding the Patched receptor (PTCH) are associated with hereditary and sporadic basal cell carcinomas and sporadic medulloblastoma. Loss of PTCH, a receptor for the ligands in the hedgehog family, leads to constitutive activation of the atypical G protein–coupled receptor (GPCR)–like protein Smoothened (Smo). Cyclopamine, a naturally derived inhibitor of Smo, and other, newer small-molecule inhibitors, have had significant activity in preclinical models lacking PTCH gene function and in clinical trials. These examples illustrate that identification of loss-of-function mutations can reveal downstream target molecules in critical cancer pathways.
Translocations are typically thought of as gain-of-function genetic events in that they act dominantly. However, they can functionally inactivate endogenous genes through the creation of dominant-negative protein fusions. For example, the PML-RARα fusion protein is believed to act, at least in part, as a dominant-negative inhibitor of endogenous RARα function. This type of molecular action can be difficult to elucidate in functional experiments. Nonetheless, it is worth remembering this possibility when evaluating proteins or genes as drug targets when they are involved in new or existing translocations.
Chromosomal-level alterations leading to loss-of-function events include homozygous deletions, heterozygous deletions, and loss of heterozygosity. Homozygous deletions are typically highly focal in their nature. When the boundaries are defined with large sample sets, in many cases, only a few genes are found in the overlapping region. For example, in the case of focal deletions on 9p, the typical minimal deleted regions in tumors such as melanoma contain only CDKN2A and CDKN2B . Heterozygous deletions are typically larger and usually involve entire chromosomes or chromosome arms. As in the case of amplifications, such large-scale broad genetic alterations make it difficult to ascertain the cancer relevance of any single gene contained within a larger region. Loss of heterozygosity (LOH) can occur as an event associated with heterozygous deletion leaving only one parental chromosome behind, which is thus homozygous. LOH can also occur when duplication of the remaining parental chromosome creates a situation known as uniparental disomy. This is significant because it may indicate preservation of a mutated allele, leading to two mutant alleles of a given gene. In the case of the tumor suppressor gene p53 , copy-neutral LOH is common.
Inactivating point mutations are a common means of gene inactivation. In this case stop codons are the most efficient mechanism for disrupting gene function. As a result, tumor suppressor gene mutations often are enriched for stop or truncating mutations. This creates certain technical and cost difficulties in sequencing genes for inactivating mutations because it is necessary to sequence all coding exons, consider sequencing of the regulatory elements governing splicing, and consider the detection of promoter mutations. As a result of these difficulties, precise clinical analysis of tumor suppressor gene mutations remains challenging even with the application of next-generation sequencing technologies.
Protein Molecular Epidemiology by Immunohistochemistry.
It is important to understand the relationship between the expression or activity of a gene product and patient outcome or other clinicopathologic data. Although establishing a relationship between protein expression and clinical data (e.g., survival) does not provide a causal link to cancer, such linkage to patient outcome likely indicates a nonrandom association with cancer behavior. Here the clinician should be careful to consider the relationship between protein expression and the state of differentiation of the cancer. Because the degree of cancer differentiation state (usually referred to as grade ) is typically already linked to patient prognosis, candidate drug targets whose protein levels vary with differentiation state may be secondarily linked to patient outcome. Thus an independent association between a protein’s expression and patient outcome should be sought in multivariate analysis. In the case of the targeted disruption of HER2 in patients with breast cancer, the association of HER2 amplification with poor outcome provided a significant motivation for the development of trastuzumab, an anti-HER2 monoclonal antibody.
The ability to perform larger-scale epidemiology analyses typically relies on archived tissue for which longer-term clinical follow-up is available. This largely dictates that target epidemiology be carried out in paraffin-embedded, fixed tissue. Here RNA and DNA can be examined by in situ hybridization (ISH), fluorescence in situ hybridization (FISH), or next-generation sequencing approaches. Historically, the most common method for interrogating human tissue samples is protein-based immunohistochemistry (IHC). These studies have been made higher throughput by the advent of tissue microarrays. Such arrays align hundreds of tumor samples on a single slide, facilitating the experimental procedures. IHC is primarily limited by the quality of the antibody reagents that are used and the care with which such reagents are subject to rigorous validation. Too often, poorly described or unvalidated antibodies are used in IHC studies after a quality assessment based solely on the observed staining pattern in tissue sections. In all cases where IHC is used, the controls should include experimental, fixed cell blocks derived from negative control cells or tissues known to lack the antigen of interest and positive control cells or tissue known to harbor the antigen of interest. These controls can typically be obtained from blocks created from cell lines where the negative controls can be generated by using short hairpin RNA (shRNA). With the development of protocols for next-generation sequencing from paraffin-embedded tissues, the use of such approaches has become more commonplace for the interrogation of archived primary patient tissues.
Functional Studies to Address Cancer Dependence
Ideally, the functional relevance of a putative cancer target should be elucidated before embarking on the lengthy process of drug discovery. The concept is simple: It is preferable to “fail” or invalidate a putative target in preclinical studies than to fail later in the clinic. Although there are multiple reasons for the failure of a given therapeutic in clinical development, ideally failure should be avoided because the drug is made against an irrelevant target. Hence one should develop a robust understanding of the science surrounding a given drug target and tie this scientific understanding to cancer-relevant processes. It is useful to consider two questions in the context of designing experiments for functional validation. First, is the putative cancer target necessary for the maintenance of a cancer-related phenotype? Second, is the putative cancer target sufficient for inducing a cancer or transformation-related phenotype? These questions are generally answered by experiments directed at inducing loss of function of the target (addressing necessity) and by experiments directed at inducing gain of function (addressing sufficiency).
Gain-of-Function Experiments.
Gain of function of a given protein typically involves overexpression of the cDNA or of an activated allele generated by mutation. The functional consequences of gene activation are assayed first in in vitro growth assays, in transformation assays, and in cotransformation experiments. Typically, gain of function in transformation assays would be assessed in murine embryonic fibroblasts, in immortalized NIH3T3 cells, Ba/F3 cells, or in Rat1a fibroblasts. In addition, the ability to transform human primary epithelial cells has been made possible through the use of telomerase and has provided a set of assays that can be used to measure the ability of a given gene to induce transformation in human cells.
Short-term validation experiments using in vivo systems have become more attractive with the advent of tissue reconstitution murine models, enabling rapid genetic manipulation. Examples of this approach include the use of murine hematopoietic cells or murine fetal hepatoblast cells. In each case retroviruses directing the expression of the relevant candidate oncogene can be introduced into the respective primary cells that are then re-introduced into host animals. As an example, mutations in JAK2 discovered in myeloproliferative syndromes recapitulate the disease entity when re-introduced in retroviral vectors into the hematopoietic reconstitution system. Similarly, the use of hepatoblasts has been useful in demonstrating that certain gene activation events can induce hepatocellular carcinoma. These systems provide some of the speed and flexibility of in vitro systems but maintain the more authentic relationship between stroma and the generated tumor cells.
Fully genetically engineered models, including transgenic mice, can be useful in studying gene activation in the specific tissue of origin depending on the extent to which promoter regulation can be used to direct gene expression to the indicated cell. Knock-in animals have also been generated in which mutant RAS alleles are re-introduced into the endogenous murine RAS gene locus. Such animals have been used to demonstrate sufficiency of K-RAS for the induction of lung adenocarcinomas, carcinomas of the pancreas and myeloproliferative disease. These engineered models also provide a stable resource for testing candidate drug compounds in highly defined mechanistically driven models.
Loss-of-Function Experiments.
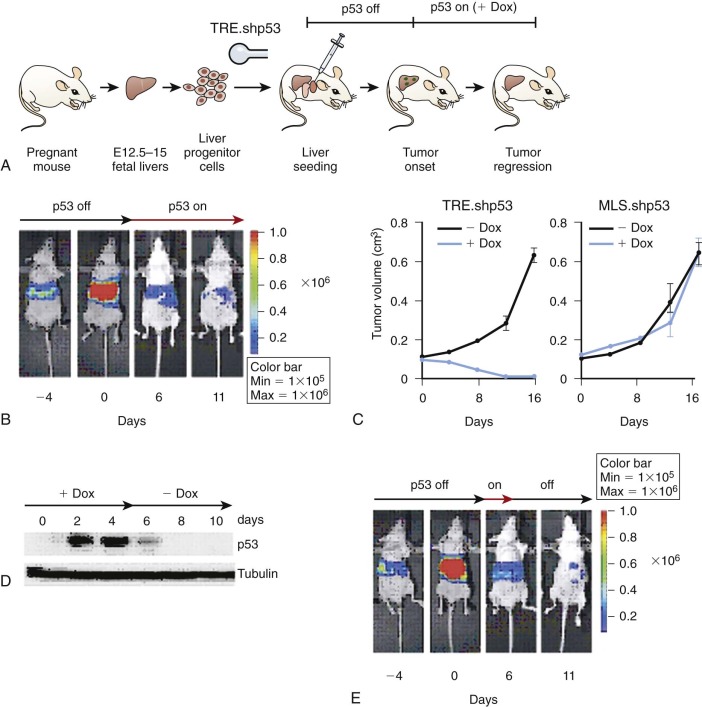
For many years there were no feasible methods for readily inducing loss-of-function alterations in mammalian systems. Consequently, the elaboration of a cell-penetrant small-molecule inhibitor or tool compound was one of the only ways to achieve the desired inhibitory effect. The development of robust small interfering RNA (siRNA) methods, taking advantage of the endogenous siRNA processing systems, now allows for transient, stable, or regulated knockdown of nearly any mammalian gene product. Genome-editing technologies, such as transcription activator-like effector nucleases (TALEN) and the CRISPR (clustered regularly interspaced short palindromic repeats)/Cas system have also been developed to enable targeted gene-disruption events. These powerful new tools are complemented by existing methods for loss-of-function experiments, including the use of inhibitory antibodies for extracellular targets, soluble protein traps for extracellular ligands and receptors, dominant-negative proteins expressed from cDNAs, murine germline knockouts, and inducible knockouts. Generally, loss-of-function experiments would be attempted first in human cancer cell line models. Increasingly, cell line models are being characterized in greater genetic detail, enabling more rational selection of models for functional inactivation experiments by either small molecules or genetic perturbations. Additionally, shRNAs have been merged with the bone marrow transplantation and the hepatoblast tissue reconstruction models described previously ( Fig. 44-2 ). Finally, regulated germline shRNAs can now be engineered in the mouse as a rapid method for generating tissue-specific and temporally regulated gene knockdowns.
Genetic Studies in Lower Organisms
The topic of genetic studies in lower organisms is too broad for a complete discussion in this chapter. However, it is worth noting the utility of defining genetic dependence in the context of highly conserved pathways that are shared between mammals, flies (Drosophila melanogaster) and worms (Caenorhabditis elegans). In some cases lower organisms have a reduced set of homologues for any given pathway member, and thus the necessity of a single gene can be tested directly. Although establishing the relationship between a gene and a cancer phenotype in these organisms is not usually possible, the genetic dependence or epistatic relationships elucidated in such model systems have proved highly informative in humans. A notable example is the discovery of the relationship between loss-of-function mutations in the tuberous sclerosis genes and activation of the mTOR and S6K (ribosomal S6 kinase) pathway (discussed later).
Target Credentialing Summary
The credentialing of drug targets ultimately must be conceived as a set of hypotheses to be tested experimentally. In each case these hypotheses must be built around the relevant biology. It is important to distinguish targets with cell-autonomous effects versus those in which the interaction between cancer cell and host factors is critical. In the case of drug targets that are encoded by bona fide human oncogenes with definitive genetic alterations, one can have a high degree of confidence in the likely therapeutic effect of inhibitory molecules. Unfortunately, this group is the minority of the drug targets for consideration. Thus the intent of credentialing is to enable a rational stratification of potential drug targets and a rational application of resources to specific drug discovery efforts.
Classes of Targeted Therapies
Targeted therapies can be divided into two main classes: drugs (small molecules, natural products, antisense, and siRNA) and biologics (antibody, protein/peptide, and cell-based) ( Box 44-6 ). Biologics, as defined by the U.S. Food and Drug Administration (FDA), include a wide range of products such as vaccines, blood and blood components, somatic cells, gene therapy, tissues, and recombinant therapeutic proteins. They can be composed of sugars, proteins, or nucleic acids or complex combinations of these substances, or they may be living entities such as cells and tissues. Biologics are isolated from a variety of natural sources—human, animal, or microorganism—and may be produced by biotechnology methods. From a practical point of view, drugs entering the market require the filing of a New Drug Application (NDA), whereas biologics require the filing of a Biologic License Application (BLA).
DRUGS
Small molecules
Natural products
Antisense oligonucleotides
RNA interference
BIOLOGICALS
Antibody
Protein
Whole protein
Peptides
Soluble receptors
CELLULAR
Drugs
Small molecules are defined as carbon-containing compounds that usually have a molecular weight of less than 2000 g/mol. These small organic compounds have the advantage of purity, high permeability into target cells, ease of manufacturing, and stability, and they remain the primary class amenable to oral delivery. However, disadvantages include insufficient selectivity resulting in off-target toxicity, challenges with poor bioavailability, and challenges with metabolism of the molecule. Over the last decade synthetic methods have been developed for producing a diversity of chemical structures, either by using directed medicinal chemistry approaches or by using combinatorial chemistry, the synthesis of numerous organic compounds by combining variations of each of the building blocks that make up the compounds. Similarly, there has been the elaboration of a diverse set of methods for identifying the initial small-molecule hits against a desired target. High-throughput screening involves the in vitro assay of target activity against chemical libraries. In silico screening can be used to dock millions of compounds into known or modeled three-dimensional structures of a given target. Fragment-based screening uses either radiographic or nuclear magnetic resonance–based structural approaches to identify very-low-molecular-weight fragments that can interrogate the binding surface of a new target. Finally, target-specific cell-based screens can be used to identify cell-penetrant small molecules perturbing a target-specific reporter or phenotype.
Natural products have been important in drug development and have played a dominant role in cancer therapy. Approximately three fourths of the current anticancer drugs are natural products or derived from natural products. The chemical diversity of natural products complements that of synthetic libraries. As a result of a long evolutionary process, natural products tend to be sterically complex with greater diversity of ring systems compared with synthetic and combinatorial libraries.
Natural products are classified according to shared scaffolding elements and can be divided into several structural classes, such as terpenes, alkaloids, polyketides, and nonribosomal peptides. The building blocks for the natural products are most often monomeric components of primary metabolic pathways that are then shunted into secondary metabolic pathways. Plants and soil microbes are traditional sources of natural products with new sources, such as fungi and marine life (e.g., sponges and algae), under active exploration.
Drug development with natural products may have specific advantages to that of synthetic molecules. The complexity of natural products may lead to greater specificity. In addition, unanticipated activities may be discovered that would be difficult to engineer into a small molecule. For example, rapamycin (derived from a soil fungus from Easter Island [Rapa Nui]), creates a novel protein-drug-protein interaction between the protein FKBP12, rapamycin, and the mTOR kinase, resulting in an exquisitely selective inhibition of mTOR. However, although the natural products have been developed into very successful drugs, there are several disadvantages to these libraries. First, natural product extracts are generally impure, and hence identifying the active compound can be difficult. Second, the synthesis of these compounds in production level quantities for medicinal purposes can be challenging. Third, subsequent optimization of the lead natural product can be difficult in the absence of a complete in vitro synthesis of the relevant natural product. New high-throughput strategies are under development to tackle roadblocks to the use of natural products for medicinal purposes.
Antisense oligonucleotides are single strands of short deoxynucleotide sequences (18-21 oligomers) that bind to target mRNA sequence by Watson-Crick hybridization and induce transcript destruction. They interact with the target transcript with sequence specificity and inhibit production of the target protein by several potential mechanisms. First, they activate endogenous nucleases, such as RNAse H, which then cleave the RNA strand of an RNA-DNA heteroduplex. A second potential mechanism of antisense activity is a noncatalytic effect with steric inhibition of the translational machinery with induction of translational arrest and inhibition of protein synthesis. A third mechanism is interference with normal RNA splicing, resulting in the inhibition of specific splice variants. Antisense therapeutics held the promise of pharmacologically inhibiting targets that had been intractable with small molecule approaches. In theory, virtually any gene can be targeted with antisense approaches with an expectation of high target selectivity. However, the development of antisense therapy has faced many challenges, including poor solubility, limited intracellular uptake, and rapid degradation. To overcome these issues, subsequent antisense therapy focused on chemical modification of the backbone to which the nucleoside bases are attached.
Despite these efforts, the utility of antisense therapy for cancer has been disappointing. However, there have been some clinical responses seen in trials. Oblimersen (Genasense), a phosphorothioate antisense therapy targeting BCL-2, has shown some activity in patients with heavily pretreated chronic lymphocytic leukemia (CLL). Furthermore, recent data from a randomized phase III trial for patients with relapsed or refractory CLL comparing fludarabine plus cyclophosphamide with or without oblimersen demonstrated an increase in complete response and nodular partial response in patients in the oblimersen arm, particularly for those patients with fludarabine-sensitive disease.
RNA interference (RNAi) is a more recent approach to nucleic acid–based therapy. Similar to antisense therapy, RNAi may have therapeutic application, particularly for targets that have been considered intractable by traditional drug discovery approaches. RNAi is a double-stranded RNA (dsRNA)–induced mechanism of gene silencing naturally occurring in plants and animals. Over the last few years, it has become possible to target virtually any gene in the genome for knockdown with small, dsRNA gene-silencing by using siRNAs or expressed shRNAs. The RNAi pathway is hypothesized to have evolved early in eukaryotes as a protection against viral and genetic pathogens. Double-stranded RNA viruses and genetic elements are subject to RNAi-dependent gene silencing as a means of cell-based immunity. Furthermore, endogenously expressed shRNAs may regulate gene expression during development through the RNAi pathway. If exploited as a potential cancer therapy, RNAi has the potential advantages of (1) relative specificity for a particular gene target, (2) efficiency with the potential to achieve more than 90% knockdown, and (3) widespread applicability with the potential of targeting any gene in the human genome. Unlike antisense oligonucleotides, RNAi takes advantage of a naturally occurring cellular mechanism for gene silencing.
As in the case of oligonucleotides, there are impasses to the development of RNAi-based therapies, including the difficulty of delivering large nucleic acid molecules to intracellular targets in humans. Unmodified siRNAs have a short half-life in human plasma and are rapidly excreted by the kidneys. Furthermore, to have activity the siRNA or shRNA must reach the cytoplasm of the targeted cell, and naked siRNAs are not efficiently taken up by mammalian cells unaided. One method of delivering shRNA that is commonly used in the laboratory to develop stable knockdown is the use of viral vectors. However, with viral vector delivery, there remains significant concern regarding insertional mutagenesis, carcinogenesis, direct cellular toxicity and short-lived on-target effects. Other nonviral methods of delivery are actively under exploration. One possibility is to chemically modify the siRNA to increase its stability with modifications such as locked nucleic acid (LNA) residues or phosphorothioates. A second approach to increase the stability of the siRNA is to package it in liposomes or other nanoparticles such as the cationic polymer polyethylenimine or to use cholesterol modifications. These packaging systems can also be modified to contain tissue-specific homing signals, such as the attachment of specific antibodies. Despite the problems with delivery and stability of RNAi, novel therapeutics using RNAi have entered clinical trials for multiple diseases.
Biologicals
Antibody therapy with monoclonal antibodies (Mabs) is an increasingly important mode of targeted therapy for cancer. Early attempts at antibody-based therapy with murine Mabs were limited by immunogenicity, short half-life in humans, and poor ability to activate human immune effector mechanisms. With the evolution of recombinant DNA technology, it is now routine to create humanized Mabs. Recombinant antibodies with human crystallizable fragments (Fc) regions are much less immunogenic in humans than murine Mabs and can facilitate activation of immune effector mechanisms. Thus recombinant Mabs may have a dual mechanism of anticancer activity secondary to the specificity of epitope targeting with the antigen-binding fragment (Fab) and the recruitment of immune effectors with the Fc region. As a direct effect of the Mab variable domain binding to the target, they can have antagonist effects on receptor-ligand interactions and receptor-receptor interactions or agonistic effects on the target’s biological activity. Second, if engineered to be of the immunoglobulin G1 (IgG1) subclass and to contain an Fc region, they induce immune effector function against I the target cell after their interactions with complement or with receptors for the Fc region, such as FcγR, a class of surface glycoproteins expressed predominantly on white blood cells. The Fc region interactions with FcγR can induce antibody-dependent cell-mediated cytotoxicity (ADCC), phagocytosis, endocytosis of immune complexes followed by antigen presentation, and release of inflammatory mediators. Antibodies can be linked to chemical moieties with additional antitumor activity, such as in gemtuzumab ozogamicin, where an antibody to CD33 was conjugated to the antitumor antibiotic calicheamicin.
Bispecific antibodies: Whereas MAbs that are clinically effective usually recruit immune cells expressing an Fc receptor, T cells are notably FcR negative. To engage T cells in the antitumor response, bispecific antibodies (bsAbs) can be deployed. These are artificially designed molecules that bind simultaneously to two different antigens, one on the tumor cell, the other one on an immune cell, such as T cells, cross-linking tumor cells and T cells. For example, blinatumomab, a bsAb that directs T cells to CD19-expressing tumor cells, such as B-ALL and non-Hodgkin lymphoma (NHL) cells, has been demonstrated to have compelling activity in clinical trials, including complete responses.
Antibody-based therapy has several advantages to small molecule–based therapy ( Box 44-7 ). First, there is a high degree of specificity for the target of interest, resulting in reduced off-target effects. Second, pharmacokinetic properties are very similar from one antibody product to another, with the half-life of an infused antibody generally lasting 2 weeks. Unlike the case of small molecules, there is a consistent path for metabolism, thus eliminating a highly variable aspect of low-molecular-weight drug development. Third, the spectrum of targets amenable to antibody-based therapeutics is largely distinct from small molecules and includes large extracellular domains and secreted ligands that are otherwise “undruggable.”
Advantages
- 1.
High-degree of specificity results in reduced off-target effects.
- 2.
Pharmacokinetic properties are similar across different antibodies.
- 3.
Spectrum of targets amenable to antibody-based therapy differs from that of small molecules.
Disadvantages
- 1.
Intravenous administration is necessary.
- 2.
Preclinical in vivo studies are difficult to perform.
- 3.
Neutralizing antibodies can develop.
- 4.
Antibodies cannot access intracellular antigens.
- 5.
Volume of distribution is limited by large molecular weight of antibody.
- 6.
The cost of production is higher.
On the other hand, there are several disadvantages of antibody-based therapy compared with small-molecule therapy. Currently antibody-based therapies are generally delivered intravenously. A second disadvantage is that preclinical in vivo studies can be difficult to conduct. The researcher must address whether the antibody in development will recognize the appropriate antigen in the animal model being tested. In some cases only primates conserve the relevant human epitope, limiting the selection of species available for toxicology studies. Efficacy studies, in particular against stromal or host factors, may require generation of a “murine equivalent” antibody, although equivalence is difficult to quantitate. The robust evaluation of host effector function elicited by an antibody and the role of this function in antitumor activity is also difficult to assess in the typical human tumor xenotransplant study in rodents. A third challenge with antibody-based therapy is that the patient can develop neutralizing antibodies to the therapy. This was particularly problematic with murine-based antibody therapy. Fourth, antibodies cannot easily gain access to intracellular antigens. Fifth, the volume of distribution of antibodies is limited by the large molecular weight, and tumor penetration is therefore a concern, at least theoretically. Finally, antibody-based treatment is generally more costly to produce compared with small-molecule therapy.
Cellular Adoptive Therapy
Most recently, cell-based biological products have also seen dramatic clinical responses. The transfer of autologous T cells modified to express chimeric antigen receptors (CARs) specific for the B-cell antigen CD19 has shown antitumor activity in low-grade B-cell malignancies and in refractory adult and pediatric B-cell ALL. These exciting advances are discussed in more detail in Chapter 46 .
Targeted Therapy: Lessons Learned from Adult Oncology
With rare exceptions, most drugs are first tested in adults before evaluation in children. This has been true for targeted agents as well. In the following section several examples of successful targeted therapies for the treatment of adult patients with cancer are briefly discussed, with an emphasis on particular lessons learned in the development and testing of these early agents.
Genetics Examples of Targeted Therapy
Treatment of Acute Promyelocytic Leukemia with All- Trans -Retinoic Acid.
The successful treatment of patients with M3-AML (acute promyelocytic leukemia, or APL) with ATRA is one of the earliest examples of targeted therapy in the genetic class. This therapeutic discovery was serendipitous. The observation that myeloid blasts differentiate in response to retinoic acid derivatives preceded the discovery of the involvement of the retinoic acid receptor in the pathogenesis of APL. ATRA treatment is one example of how discovery of a compound inducing a phenotypic alteration can lead to greater understanding of the mechanisms underlying the state change. By morphologic examination APL is characterized by a predominance of malignant hypergranular cells blocked at the promyelocyte stage of differentiation. At the molecular level a reciprocal translocation involving the retinoic acid receptor α (RARα) gene on chromosome 17q21 is invariably present. This translocation most commonly fuses RARα to the PML gene on chromosome 15q22. RARα is a hormone-dependent, DNA-binding, nuclear receptor transcription factor that can act as a transcriptional activator or inhibitor. In the presence of physiologic amounts of retinoic acid, it normally functions as a transcriptional activator. Abnormal RARα fusion proteins function as transcriptional repressors, enhancing interactions with the corepressor complex. Pharmacologic doses of ATRA appear to overcome the PML-RARα−induced transcriptional repression by dissociating the corepressors from PML-RARα and restoring normal ATRA-mediated myeloid differentiation. Furthermore, the PML-RARα protein undergoes proteolytic cleavage in APL cells after ATRA treatment. ATRA treatment increases the fraction of differentiated cells with functional characteristics of neutrophils, induces a mature membrane phenotype, inhibits leukemia cell proliferation, and ultimately induces apoptosis. Historically, APL was among the most fatal subtypes of AML. However, with the addition of ATRA to APL therapy regimens, the overall 2-year survival rate is more than 75%. APL now has the highest cure rate of the AML subtypes.
The success of ATRA in the treatment of PML-RARα rearranged AML illustrates several important issues regarding targeted therapy. First, specific somatic rearrangements, such as PML-RARα, can be exploited with targeted therapy, and the specific underlying genetic event predicts response, not the phenotype. For example, other RARα rearrangements, such as PLZF- RARα, are associated with an APL phenotype. However, the PLZF-RARα fusion protein is associated with nuclear corepressor interactions that are resistant to ATRA therapy. Patients with the PLZF-RARα rearrangement are resistant to ATRA therapy. A second important concept that emerges from the success of ATRA is that differentiation therapy is feasible for some malignancies. In general, terminally differentiated cells lose the capacity to divide and ultimately undergo apoptosis. This is indeed the case for APL cells treated with ATRA, which raises the possibility that such approaches may be more broadly applicable. A third important lesson from this example is that modulation of transcription factors is therapeutically feasible. Rearrangements involving transcription factors are a common event in the acute leukemias and in the pediatric solid tumors. Although well-characterized contributors to the malignant state, these transcription factor abnormalities have been considered “undruggable” or pharmacologically intractable because there is no obvious approach to inhibiting their function. However, ATRA therapy for the genetic lesion PML-RARα lends credence to the idea that these lesions can be targeted and that their modulation can have therapeutic efficacy. Finally, this example reinforces the importance of combination therapy not only for cytotoxic agents but also for targeted therapy. Although the majority of patients with APL in early trials achieved a complete remission with ATRA as a single agent, it was only in combination with conventional chemotherapy or arsenic trioxide that long-term cure rates were achieved.
Imatinib in Chronic Myelogenous Leukemia.
The development of imatinib for the treatment of CML stands out as the first example of a designed small-molecule inhibitor developed for the purpose of targeting an activated oncogene. Moreover, its dramatic single-agent activity in its initial phase I and II trials, along with the rapid path to registration, has provided the impetus for attempting to identify highly responsive patient populations during preclinical and early clinical development. In 1960 Nowell and Hungerford first detected the presence of the then-named Philadelphia chromosome in nearly all patients with CML. This aberrant chromosome, resulting from the fusion of chromosomes 9 and 22, was subsequently shown to encode a fusion protein linking a gene of unknown function ( BCR, or breakpoint cluster region ) to the product of the Abelson gene—the cellular homologue of an oncoprotein activated by the Abelson leukemia virus. The discovery that the Abelson gene product (ABL) could function as a protein tyrosine kinase and that the normal autoregulatory domain of ABL was replaced with coding sequences from BCR led to the idea that constitutive activation of ABL kinase activity was likely a causal genetic event in CML. In animal models production of BCR-ABL was sufficient to induce both acute and chronic leukemia. These data, along with the presence of a catalytic domain, made BCR-ABL kinase an attractive drug target. Based on these elements, a drug-discovery program was launched, culminating in the discovery and clinical testing of imatinib (Gleevec), a small molecule inhibitor of BCR-ABL.
In the initial phase I trial by Druker and colleagues, 83 patients with interferon-refractory CML were enrolled and treated with doses of imatinib ranging from 25 to 1000 mg per day. No MTD was reached, and nausea, myalgias, edema, and diarrhea were the most frequent side effects. Remarkably, at a dose of 140 mg or higher, all patients had a hematologic response. At doses of 300 mg per day or greater, 53 of 54 patients had complete hematologic responses, 17 had major cytogenetic responses, and 7 had complete cytogenetic remissions. Numerous subsequent trials demonstrated remarkable activity in CML and CML in accelerated phase, lymphoid or myeloid blast crisis or with Ph+ ALL in adults, including complete molecular responses.
On the basis of these data, imatinib became the gold standard for first-line therapy of CML. Nonetheless, resistance to imatinib does occur. In vitro experiments conducted by using randomly mutagenized ABL cDNA libraries inserted into sensitive CML cells revealed that a defined spectrum of mutations in the ABL gene itself could preserve kinase activity but could disrupt binding to imatinib. More important, sequencing of the ABL kinase domain in patients relapsing on imatinib revealed a similar spectrum of mutations in patient samples. When these mutations were isolated and placed into ABL kinase constructs and re-inserted into sensitive CML cells in vitro, these mutant ABL constructs also rescued lethality induced by imatinib inhibition. These data, although initially concerning because of the potential for long-term control of the CML disease state, definitively demonstrated in humans the absolute requirement for ABL kinase activity in CML and closed the loop on the notion of a fully validated drug target. Moreover, the identification of such mutants provided the impetus for testing second-generation inhibitors that might overcome these resistance alleles. Here, two separate approaches were considered. First, although imatinib is a fairly selective inhibitor, it is not a particularly potent inhibitor. Because many mutations shifted the IC 50 of the kinase activity, it remained possible that a more potent inhibitor might be able to overcome or prevent the emergence of these resistance alleles. Nilotinib is a second-generation inhibitor designed to target BCR-ABL and preserve the interaction with the inhibited form of the ABL kinase, the type II binding mode. Nilotinib is more potent in biochemical and cellular assays of ABL activity and in phase I and phase II trials had significant activity in imatinib-resistant CML. Nilotinib was later demonstrated to be superior to imatinib in patients with newly diagnosed chronic phase CML. Dasatinib, initially developed as a src kinase inhibitor, binds to the “active” form of its kinase targets. Src kinases are highly related to ABL kinase activity, and in vitro testing showed that dasatinib had activity against imatinib-resistance alleles. In phase I and II trials dasatanib has shown significant activity in imatinib-resistant patients. Both dasatinib and nilotinib have gained FDA approval in imatinib-resistant patients with CML and in CML-accelerated phase and blast crisis. Ultimately, both drugs were approved for upfront therapy of patients with CML.
Unfortunately, both nilotinib and dasatinib lack activity against a common but difficult to target ABL mutation, the T315I or “gatekeeper” mutation. This mutation creates a steric problem in the adenosine triphosphate–binding domain. This particular kinase domain conformation creates challenges in the drug design process. Notably, it has been difficult to make potent and selective inhibitors of this conformation inhibition. Recently, a drug with activity against the T3151 mutation, ponatinib, was granted accelerated approval by the FDA for the treatment of adult patients with chronic, accelerated, or blast-phase CML that is resistant or intolerant to prior tyrosine kinase inhibitor therapy or BCR-ABL–positive ALL that is resistant or intolerant to prior tyrosine kinase inhibitor therapy ( Fig. 44-3 ). However, the safety of this drug was later called into question secondary to concerns about drug-related arterial thrombotic events.
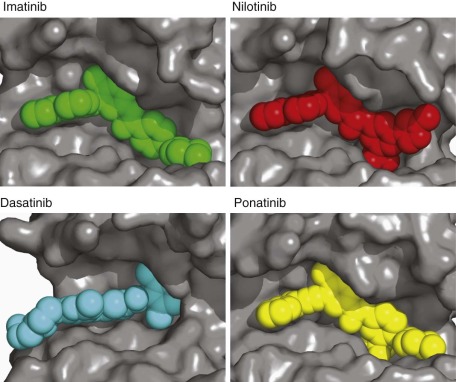
Imatinib and Other Malignancies.
Imatinib, in addition to inhibiting ABL kinase activity, also inhibits c-KIT and platelet-derived growth factor receptor (PDGFR). In preclinical cell line models of gastrointestinal stromal tumors (GIST), a tumor defined by frequent mutations in the c-KIT gene, efficacy of imatinib both in vitro and in murine xenograft models was demonstrated. Based on these early preclinical findings, a patient with inoperable, recurrent, and treatment-refractory c-KIT -mutated GIST was treated with imatinib. After 4 weeks of therapy, the fludeoxyglucose-positron emission tomography scan showed a complete resolution of tracer uptake compared with the pretreatment scan, and over the ensuing 8 months the patient had a progressive diminution in the tumor volume. Multiple subsequent studies demonstrated response to tyrosine kinase inhibitors in GIST, and in 2008 the FDA granted accelerated approved for imatinib in the adjuvant setting for completely resected primary GISTs 3 cm and larger.
The activity of imatinib against PDGFR or KIT has enabled clinical development in other diseases characterized as harboring specific genetic alterations activating these receptors. In the case of dermatofibrosarcoma (DFS) the majority of patient lesions harbor translocations between chromosomes 17 and 22 fusing the PDGFB ligand gene downstream of the Col1a promoter. This leads to constitutive ligand production and receptor activation and sensitivity to imatinib in in vitro models, transplantable primary tumors from patients, and in patients treated with the drug. In hypereosinophilic syndrome (HES) Schaller and Burkland report that empiric imatinib treatment of a patient with HES induced a complete and rapid remission of the disease. Similarly, other studies revealed patients with HES responding to imatinib. Gleich and colleagues reported the empiric treatment of five HES patients, four of whom had a prompt and sustained response to imatinib with a resolution of the eosinophilia. The basis for these dramatic observations was uncovered in 2003, when Cools and coworkers reported the discovery of an intrachromosomal deletion that placed PDGFRA downstream of the promoter of the FIP1L1 gene, resulting in constitutive upregulation of the receptor.
The remarkable finding in the examples of the FDA-approval of imatinib for CML, GIST, dermatofibrosarcoma, and HES is that in each case a specific genetic event leads to constitutive activation of a molecular target of imatinib, and in each case a high degree of efficacy is achieved. Moreover, we have learned that efficacy is identified rapidly in selected patient populations, with dramatic responses even in phase I. We have learned that MTD does not necessarily set the appropriate effective dose, and, indeed, MTD end points were not reached in a number of the early phase I trials.
Dramatic Responses in Select Genotypes.
With the success of imatinib for the treatment of patients with CML, a high bar was set for subsequent small-molecule kinase inhibitors. The possibilities that this targeted approach held for the poorly treated adult epithelial malignancies generated great excitement. The EGFR pathway is believed to be important in the development, maintenance, and proliferation of many malignancies, and EGFR is frequently overexpressed in epithelial solid tumors. For these reasons EGFR was an attractive target for small-molecule inhibition. Gefitinib (Iressa) and erlotinib (Tarceva), both orally administered tyrosine kinase inhibitors, were developed to bind competitively at the adenosine triphosphate pocket of EGFR, inhibiting autophosphorylation, and suppressing downstream EGFR signaling. Surprisingly, these EGFR inhibitors have been largely ineffective in the treatment of solid tumors with one exception: the treatment of a subset of patients with non–small cell lung cancer (NSCLC).
After these initial clinical studies, numerous reports characterized the clinical predictors of tumor response (as opposed to survival) in patients. It soon became apparent that these patients frequently shared similar demographic features: a nonsmoking history, a well-differentiated adenocarcinoma histology, female sex, and Asian origin, although the underlying genetic reason for these response differences was not known. One hypothesis was that expression levels of EGFR correlated with these clinical characteristics and hence response. However, these initial clinical trials failed to identify a correlation between receptor expression and clinical outcome.
Later, in 2004, two research groups independently identified a correlation between mutations in the tyrosine kinase domain of the EGFR and patient responders. These EGFR mutations enhance ligand-dependent EGFR activation and sensitivity to gefitinib/erlotinib and are more common in nonsmokers, women, Asians, and patients with adenocarcinoma. Approximately 90% of these mutations occur in a few amino acids. Between 45% and 50% of mutations are in-frame deletions of exon 19 (codons 746 to 750), and 35% to 45% of mutations are missense mutation leucine to arginine at codon 858 (L858R) in exon 21. Several studies have also reported that patients with EGFR mutant tumors have a longer survival when treated with EGFR small- molecule inhibitors compared with patients with wild-type EGFR tumors.
This work highlights the importance of developing a molecular understanding of disease and response. The molecular characterization of EGFR abnormalities in NSCLC and their relationship to response to tyrosine kinase inhibitors have been illuminating for the clinical application of targeted therapy. More recently, the dramatic responses seen to B-RAF inhibition in patients with V600E B-RAF -mutant melanoma and to ALK inhibition in patients with EML4-ALK rearranged lung cancer underscores the critical role of matching tumor genotype with targeted drug therapy ( Fig. 44-4 ).
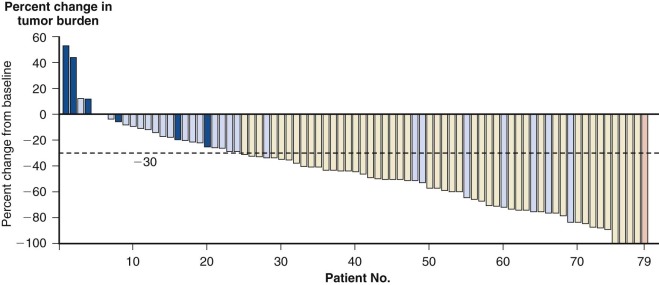
Lineage-Dependence Examples of Targeted Therapy
Several successful targeted cancer therapies capitalize on the lineage dependence of the tumor as exemplified by antibody-based therapy directed against cancers with hematologic lineage markers such as rituximab (Rituxan). As discussed in the subsequent sections, these therapies have the advantage of target specificity, with predictability of side effects based on the expression pattern of the lineage marker.
Rituximab Therapy for Non-Hodgkin Lymphoma.
Rituximab (Rituxan) in 1997 became the first monoclonal antibody approved by the FDA for the treatment of malignancy (NHL). It is a chimeric, anti-CD20 monoclonal antibody composed of murine variable regions fused with human IgG1 heavy and κ light chains. CD20 is a transmembrane protein expressed on normal and malignant mature B lymphocytes thought to be important in calcium transportation and to be involved in B-cell activation, differentiation, and growth regulation. Rituximab binds to CD20 and depletes CD20-positive B cells in the circulation and in lymph nodes.
The first trials evaluating rituximab were in patients with indolent lymphoma, which has no curative therapy. The pivotal multicenter trial in patients with relapsed low-grade or follicular lymphoma demonstrated an overall response rate of 50% and a complete remission rate of 6%. These results were comparable with the best single-agent cytotoxic therapy for relapsed follicular lymphoma. Rituximab has also been evaluated in combination with cytotoxic chemotherapy for patients with indolent B-cell lymphomas. One of the earliest studies, published in 1999, evaluated 40 patients with low-grade or follicular lymphoma treated with six cycles of CHOP ( c yclophosphamide, h ydroxydaunorubicin, o ncovin, and p rednisone or p rednisolone) and rituximab (R-CHOP). The initial report suggested an additive benefit of rituximab with no additional toxicity when these two therapies are used in combination. The overall response rate was 95%, and the complete response rate was 55% in the intent-to-treat group. One of the larger clinical trials of 428 patients with previously untreated, advanced-stage follicular lymphoma assigned to CHOP or R-CHOP demonstrated an overall response rate of 90% with CHOP and 96% with R-CHOP and estimated survival rate at 2 years of 90% and 95%, respectively.
The combination of rituximab with cytotoxic chemotherapy has also been evaluated for aggressive NHL. In a randomized phase III study comparing CHOP with R-CHOP in elderly patients with diffuse large B-cell lymphoma, the rate of complete response was higher in the group receiving R-CHOP (76% versus 63%, P = 0.005). Furthermore, at 2 years both event-free and overall survival rates were higher in the R-CHOP arm, with no significant additional toxicity. A randomized study of CHOP-like chemotherapy alone or in combination with rituximab in adult patients ages 18 to 60 years with good-prognosis diffuse large B-cell lymphoma demonstrated a 3-year event-free survival rate of 59% with chemotherapy alone versus 79% with the addition of rituximab and at 6 years of 56% and 74%, respectively.
Thus R-CHOP is now considered the standard of care for the first-line treatment of CD20-positive diffuse large B-cell lymphoma and for CD20-positive follicular B-cell NHL in combination with chemotherapy. Rituximab is also indicated for the treatment of patients with relapsed or refractory CD20-positive low-grade or follicular NHL.
Host Track Example of Targeted Therapy
Bevacizumab and Colorectal Cancer.
Angiogenesis plays an important role in tumor formation. Continued tumor growth is dependent on new blood vessels for the supply of oxygen and critical nutrients. Moreover, certain somatic and germline alterations, including those disrupting the function of the von Hipple-Lindau (VHL) tumor-suppressor gene, appear to act largely by triggering dysregulated hypoxic and angiogenic responses. Over the last several years, numerous angiogenic growth factors have been characterized, including vascular endothelial growth factor (VEGF). VEGF is a member of a large family of dimeric glycoproteins acting as growth factors. It is essential for the normal development of blood vessels and is a growth factor for vascular endothelium. VEGF binds to the transmembrane tyrosine kinase receptors VEGFR1, VEGFR2, and VEGFR3. In the malignant state VEGF secreted by the tumor or surrounding stroma leads to endothelial cell proliferation and migration. Because VEGF and its receptors play a critical role in tumor formation, progression, and maintenance, VEGF or VEGFR inhibition became a focus of targeted antiangiogenesis therapy. Several approaches are in development, but the two with the most success thus far are (1) neutralizing antibodies that inhibit the binding of VEGF to its receptors and (2) small-molecule tyrosine kinase inhibitors that block downstream signaling from the VEGFR.
Bevacizumab (Avastin) is a recombinant, humanized monoclonal antibody that binds to VEGF-A, preventing its interaction with the VEGF receptors. It is believed that bevacizumab affects tumor vasculature by several mechanisms: (1) inducing regression of tumor vasculature, (2) normalizing the tumor vasculature, (3) inhibiting new blood vessel formation, and (4) preventing recruitment of progenitor cells from the marrow. It may also improve chemotherapy delivery by altering tumor vasculature and decreasing interstitial pressure. Bevacizumab has been approved by the FDA for first- (2004) and second- (2006) line treatment of patients with metastatic colorectal cancer and the treatment of nonsquamous metastatic lung cancer, although it extends survival by less than 6 months.
As with most of the targeted therapies, many of the side effects of bevacizumab were predictable. Some of the reported side effects include hypertension, proteinuria, arterial thromboembolic events, bleeding, poor wound healing, gastrointestinal perforation, and reversible posterior leukoencephalopathy syndrome (RPLS). Most of these side effects are predictable from the known activity of VEGF. For example, VEGF is a homeostatic factor for regulating blood pressure. Blocking of VEGF or VEGFR leads to increased vascular tension and thus problems with hypertension and RPLS. It is interesting to note that the side effects of VEGF inhibition are similar to the signs of preeclampsia and eclampsia (hypertension, proteinuria, and mental status changes), a disease in which soluble VEGFR-1, which antagonizes VEGF functions, has been implicated.
Other VEGFR pathway inhibitor development has focused on inhibition of the kinase activity of the VEGF receptor. In December of 2005 the FDA approved the drug sorafenib (Nexavar), a small-molecule VEGFR inhibitor, for the treatment of patients with advanced renal cell carcinoma, and in January of 2006 the FDA approved the multitargeted inhibitor (including VEGFR and KIT) sunitinib (Sutent) for the treatment of patients with both advanced renal cell carcinoma and for patients with GIST who are intolerant of, or who have had disease progression with, imatinib treatment.
The “success” of bevacizumab for colorectal cancer is a humbling one for drug development. In reality, there is a long way to go when the successful drug extends overall survival by less than 6 months. In pediatric cancer care this would hardly be deemed a victory. However, many lessons have been learned from this experience. One is that antiangiogenesis-directed therapy can play a role in some disease treatments. A second is the continued importance of combination therapy, even for targeted therapy. Bevacizumab alone had very little activity. It was in combination with standard cytotoxic agents that a clinical benefit was demonstrated. Third, careful evaluation of targeted therapy benefits and toxicities may provide new insight into human disease.
Empiric or Synthetic Lethal
As described previously, classical chemotherapy is likely to exploit cancer-dependent states that are not fully understood. For example, microtubule inhibitors, such as the taxanes, specifically interact with protein components of microtubules and are efficacious in a number of cancers, although there is no current scientific rationale for their selective efficacy. A number of emerging targeted therapeutics target highly selective protein targets involved in diverse basic cellular processes and were elaborated according to specific synthetic lethal hypotheses.
Histone Deacetylase Inhibitors.
Histone deacetylase (HDAC) inhibitors are an example of an empirically acting targeted agent. Transcriptional repression and transcriptional activation, at least in part, reflect the balance of histone modification of chromatin structures surrounding gene regulatory regions. Histone acetylation is of particular importance. In many cases transcriptional activators recruit histone acetylase activity to gene regulatory regions, whereas transcriptional repression complexes typically recruit HDAC to the same regions. In keeping with the notion that repression might underlie the function of oncogenic transcription factor translocations, HDAC enzymes are recruited to the critical promoters by various translocation partners, including RARα and ETO. These data provide a compelling rationale for the development and testing of HDAC inhibitors. In keeping with the underlying concept of synthetic lethality, it is possible to hypothesize an enhanced dependence on, or constitutive requirement for, HDAC enzymes by the aforementioned repressive oncogenes. Although the original therapeutic concept evolved from a link between leukemogenesis and transcriptional repression, a large array of proteins are now known to be modulated through acetylation at the hands of 11 related HDACs. This will invariably create difficulty in elucidating the underlying therapeutic mechanisms linked to the anticancer activity now observed in the clinic with relatively broad-acting HDAC inhibitors.
An array of HDAC inhibitors targeting the major classes of HDAC enzymes are now in the clinic. The majority of these inhibitors have a core hydroxamate structure that sits deep in the enzymatic pocket, making contacts with a key coordinated zinc atom in the enzyme. The most advanced of these compounds, vorinostat (suberoylanilide hydroxamic acid [SAHA]), was approved for the treatment of cutaneous T-cell lymphoma (CTCL). In initial phase I studies testing vorinostat, diarrhea, anorexia, dehydration, and fatigue were found to be the limiting side effects. Thrombocytopenia was also frequent. Based on early evidence of efficacy in patients with CTCL, a phase II trial of vorinostat was conducted exploring three dose levels. Eight of 33 patients achieved a partial response. A phase IIB multicenter trial was subsequently conducted. In this trial of 74 patients who had all progressed or relapsed on at least two prior treatment regimens, 30% achieved an objective response and 77% of patients had some measure of disease improvement. One patient achieved a complete response. These data provided the basis for the FDA approval of vorinostat for CTCL in 2006.
Romidepsin (depsipeptide, FR901228, FK228), a natural product bicyclic depsipeptide originally isolated from a broth culture of Chromobacterium violaceum, was discovered to have potent transcriptional activation properties and to be a potent inhibitor of HDAC enzymatic activity. In the early clinical experiences with romidepsin, in addition to the observed toxicities with HDAC inhibitors discussed previously, cardiac abnormalities were also reported. In a phase I trial conducted in pediatric patients taking romidepsin, the dose-limiting toxicities were T-wave inversions and transient sick sinus syndrome, which were asymptomatic. In phase II trials romidepsin was tested in patients with neuroendocrine tumors and in patients with metastatic renal cell carcinoma. The neuroendocrine study was terminated because of serious cardiac adverse events, including ventricular arrythmias and prolonged QTc. In the renal cancer trial, prolonged QTc interval (two), grade 3 atrial fibrillation and tachycardia (one), and sudden death (one) were observed. There was insufficient antitumor activity to warrant further development in these indications. A phase II trial of romidepsin in patients with CTCL reported an overall response rate of 38%. In 2009 the FDA approved romidepsin for CTCL in patients who have received at least one prior systemic therapy. Similarly, panobinostat (LBH589), a synthetic hydroxamic acid–based small-molecule inhibitor that was specifically designed as part of a drug-discovery effort directed at producing a pan-HDAC inhibitor, has been demonstrated to have activity in patients with CTCL but also to have the potential liability of cardiac toxicity.
It is clear from the summation of these emerging data that HDAC inhibition results in significant activity in CTCL, although the reason for this empiric observation remains obscure. HDAC inhibitors are now being tested more widely in other solid tumor and hematologic malignancies. The key to these studies will be the elucidation of effective yet well-tolerated dosing regimens.
New and Emerging Opportunities in Pediatric Cancer
Targeted therapies have been introduced into the pediatric cancer therapeutic arsenal. Thus far, nearly all of these agents have already been tested in the adult community. Several have had success in the pediatric setting where the potential target is shared by the phenotypically identical adult and pediatric tumor, such as ATRA for children with APL, imatinib for BCR-ABL–rearranged CML and ALL, and rituximab for lymphoma. There are many targets—those shared by both adult and pediatric malignancies and targets specific to pediatric cancers—not yet thoroughly explored with targeted approaches. In the following sections we will discuss these new and emerging targets and the special considerations in bringing targeted therapy to the pediatric setting ( Box 44-8 ).
- 1.
Most children with cancer are cured.
- 2.
Pediatric cancer is rare.
- a.
Market incentive is reduced.
- b.
Clinical trial design and accrual are difficult.
- c.
Limited number of agents can be reasonably tested
- a.
- 3.
Toxicity may be specific to developmental stage.
- 4.
Ethical considerations are more difficult.
- a.
Informed consent for experimental therapy must be obtained.
- b.
Informed consent for participation in biological study must be obtained.
- a.
Special Considerations in Targeted Therapy Development for Children
Majority of Children with Cancer Are Cured
There are numerous special considerations in the development of targeted therapy for pediatric cancer. In contrast to adults with cancer, the majority of children with cancer will be cured of the malignancy, making the selection of pediatric patients for testing of new targeted therapies more challenging and raising the required level of activity necessary for “success.” For example, in de novo pediatric pre–B-cell ALL, with cure rates greater than 80%, it is difficult to introduce a new targeted agent for fear of detracting from an already excellent cure rate by diluting curative agents. Although there are many interesting new targets emerging, such as PAX5 and other genes important in B-cell developmental pathways, it would be difficult to justify the inclusion of upfront novel targeted therapy in patients with this excellent outcome. Thus it is likely in the relapsed setting, or in de novo disease with poor outcome, that novel targeted therapies will undergo initial testing. If efficacy and safety are demonstrated, researchers may then consider incorporation into upfront clinical trials.
Pediatric Cancer Is Rare
A total of 1,660,290 new diagnoses of cancer and 580,350 cancer deaths are projected to occur in the United States in 2013. In contrast, it is estimated that only 14,023 children in the United States younger than age 20 years were diagnosed with cancer in 2009, and 1964 died of cancer in 2009. Hence pediatric cancers are quite rare compared with adult malignancies, and there is a reduced market incentive for pediatric-specific targeted drug development by the pharmaceutical industry. Furthermore, researchers have been understandably hesitant to test new drugs in children. For these reasons, there are virtually no drugs that have been developed specifically for pediatric malignancies. Even when a common target has been identified in adult and pediatric cancer, it may be years before a drug reaches a pediatric trial. Because pediatric cancers are rare, even with a drug in hand to test, clinical trials are difficult. A rare disease incidence means slow accrual of patients and necessitates large cooperative groups to power the trial with appropriate patient numbers. The limited number of patients also precludes testing of all possible new agents. New methods are needed to more rationally choose novel targeted agents for testing in children.
One national attempt to prioritize the selection of new agents for testing in children with cancer was the development of the Pediatric Preclinical Testing Program (PPTP) by the National Cancer Institute. This group is prospectively testing new agents against a panel of xenografts and genetic models of pediatric cancer, such as neuroblastoma, rhabdomyosarcoma, osteosarcoma, and ALL. The PPTP builds on earlier work demonstrating the ability of preclinical testing of new agents in rhabdomyosarcoma and neuroblastoma xenografts to predict in vivo activity in children with these malignancies. The development of new genetically engineered models of pediatric cancer should also facilitate preclinical evaluation of targeted therapies and better prioritization of new agents for clinical trial, particularly for diseases such as Ewing sarcoma, for which no such model yet exists.
Developmental Considerations in Targeted Therapy for Children
Children may tolerate targeted therapies differently from adults secondary to age-specific differences in physiology that result in altered pharmacokinetics or to drug interference with critical developmental pathways. In normal development, vascularization of cartilage is prominent during embryogenesis and during periods of growth when neoangiogenesis occurs at growth plates. Inhibition of VEGF signaling has been reported in animal models to result in thickening of the epiphyseal growth plates secondary to an expansion of the hypertrophic chondrocyte zone. These findings have been attributed to a delayed vascular invasion of the epiphyseal growth plate with a subsequent reduced rate of chondrocyte apoptosis. Inhibition of VEGF signaling has also been reported to impair trabecular bone formation, induce dental dysplasia, and cause ovarian atrophy. Although the animal data are concerning, the actual long-term effects of this class of drug on human children is unknown.
Ethical Considerations in Targeted Therapy for Children
The testing of drugs in children raises issues universal to cytotoxic and targeted therapies, as well as challenges more specific to targeted treatments. Adults can make informed decisions about the risk and potential benefit of experimental therapies and biological studies and actively participate in treatment decisions. Particularly for young children, however, true informed consent from the patients for experimental therapies cannot be obtained. Here it is the parent or legal guardian making decisions about participation in a trial. In light of this potential conflict of interest, special regulations have been instituted to afford additional protection to children. These include the Code of Federal Regulations, Part 46; Protection of Human Subjects, Subpart D; and the related FDA regulation, Part 50, Protection of Human Subjects, Subpart D. These regulations outline allowable research in children based on the risk to the child, potential benefit to the child or other children, and alternative therapies. In the testing of targeted therapies, pharmacodynamic studies with sampling of tumor tissue before and after therapy initiation become more critical. For obvious reasons these studies can be difficult to perform with the current regulations, particularly for solid tumors or brain tumors. In general, tissue collection in children for the purpose of biological studies must be considered no greater than a minor increase in minimal risk. Innovative methods are needed to evaluate pharmacodynamic response, such as new imaging technologies, and better methods are needed to assess surrogate tissues.
Emerging Genetic Targets in Pediatric Cancer
Significant progress has been made in our understanding of the pediatric leukemias and solid tumors. As such, numerous potential genetic targets have emerged over the past 10 years. In light of the excellent cure rates for some pediatric malignancies, it might be difficult to incorporate target-directed therapies associated with good outcome genetic lesions, such as TEL-AML1 in pediatric ALL. In these early stages of targeted therapy for pediatric malignancies, the focus will likely be on lesions associated with poor outcome. In the following section we will discuss some of these genetic lesions with potential for therapeutic targeting.
FLT3 and the Acute Leukemias
Although much progress has been made in the treatment of childhood leukemias, patients with AML with mutations in the Fms-like tyrosine kinase 3 (FLT3) gene remain a high-risk group. FLT3 is a class III receptor tyrosine kinase that plays a critical role in normal hematopoiesis and shares structural homology with PDGFR, c-KIT, and CSF1R. Upon the binding of FLT3 ligand, wild-type receptors dimerize and become activated by phosphorylation, leading to activation of downstream signal transduction pathways. Mutations in FLT3 can be identified in blasts from 30% of adult patients with AML with constitutive activation of the kinase resulting from internal tandem duplications (ITD) in the juxtamembrane domain or point mutations in the activation loop. The FLT3 ITD is present in 15% and FLT3 point mutations in 7% of pediatric AML, and children with FLT3 ITD have a particularly poor prognosis.
FLT3 has also been implicated as a target in pediatric patients with MLL -rearranged ALL. Gene-expression profiling studies comparing MLL -rearranged ALL with all other ALL and AML have demonstrated a unique expression profile for MLL -rearranged leukemia. FLT3 is the gene most frequently overexpressed in MLL -rearranged leukemia compared with the other acute leukemias. An initial study reported mutations in FLT3 in approximately 15% of samples of MLL -rearranged ALL. All of these were point mutations in the activation loop, including mutations not previously reported in patients with AML (deletion of isoleucine 836). These mutations lead to constitutive activation of the kinase and increased sensitivity to small-molecule inhibitors of FLT3. Furthermore, MLL -rearranged lymphoblasts with high levels of wild-type FLT3 were sensitive to the FLT3 inhibitor PKC412. Subsequent studies have reported an incidence of FLT3 mutations in infant MLL samples ranging from 3% to 18%. These studies also confirmed increased expression of FLT3 in MLL -rearranged infant ALL compared with both infant and noninfant patients with germline MLL . Even in the absence of documented mutation, a high level of FLT3 expression was associated with FLT3 ligand independence and with increased sensitivity to the FLT3 inhibitor PKC412. In addition to its role as a target in MLL -rearranged ALL, FLT3 is a potential target in children with T-cell ALL (T-ALL), particularly with early precursor T-ALL, and in children with hyperdiploid ALL, wherein FLT3 mutations have been reported.
Several FLT3 inhibitors have been tested in clinical trials for adults with AML. Most of the early trials testing these compounds have had some transient responders with decreased peripheral blood or marrow blasts, and the compounds have generally been well tolerated. However, very few patients enter a complete remission, and the majority of patients eventually progress. The high rate of initial response failure, as well as the development of progressive disease despite an initial clinical response, has tempered enthusiasm for FLT3 inhibitors in AML. There are several possible explanations for the lack of dramatic clinical responses, including issues related to pharmacology and the failure to achieve sustained, complete inhibition of FLT3. Another explanation is the acquisition of resistance mutations in FLT3 itself. Indeed, the newer drug quizartinib (AC220), with greater potency and sustained inhibition of FLT3, has had promising initial results in clinical trials. Moreover, point mutations within the kinase domain of FLT3 -ITD, conferring quizartinib resistance, were recently reported. This finding is similar to the emergence of BCR-ABL –resistance mutations in patients with CML treated with imatinib. Significantly, this study provided gold-standard validation that FLT3-ITD is an oncogenic driver of AML.
The clinical study of FLT3 inhibitors in children with leukemia is ongoing. A phase I trial testing the multikinase inhibitor sorafenib, with activity against FLT3, in children with refractory solid tumors and leukemia, was reported by the Children’s Oncology Group (COG). This trial established a recommended phase II dose of sorafenib for children with leukemias. Rash, diarrhea, and liver enzyme elevation were the most common sorafenib-related toxicities. Two patients with AML with the FLT3-ITD achieved less than 5% bone marrow blasts with 4 or 5 cycles of sorafenib. A second phase I trial evaluated the effects of sorafenib in combination with clofarabine and cytarabine in children with relapsed or refractory leukemia. On day 8 of this trial, 10 of 12 patients demonstrated a decrease in blast percentage after sorafenib alone. After combination chemotherapy, six patients (three of these with FLT3-ITD) achieved a complete remission, two with FLT3-ITD had a complete remission with incomplete count recovery, and one patient with FLT3 wild-type had a partial remission. This trial also demonstrated downregulation of AKT, RPS6, and 4E-BP1 phosphorylation in leukemic blasts. Finally, there was another report of three patients with relapsed FLT3-ITD AML who achieved a sustained remission with sorafenib in combination with other cytotoxic chemotherapy. Based on these encouraging initial results, there are now multiple ongoing trials to test the efficacy of FLT3 inhibitors in pediatric patients with leukemia.
JAK Mutations in Pediatric Leukemia
With the marked success of imatinib in the treatment of BCR-ABL -rearranged ALL in children ( Fig. 44-5 ), there has been interest in the identification of other kinase abnormalities in childhood cancers, including leukemia. Recurrent mutations in Janus kinases (JAKs) have emerged as immediately targetable in a subset of pediatric patients with leukemia. JAKs phosphorylate substrates, including STAT proteins, on ligand binding to a type I cytokine receptor, leading to altered transcription of growth-enhancing and anti-apoptotic genes. Interest in targeting JAK2 was sparked by the finding of recurrent JAK2 mutations (most frequently involving valine 617) in patients with myeloproliferative disorders. Subsequently, activating mutations in JAK2 and JAK3 were reported in patients with AML and Down syndrome–associated acute megakaryoblastic leukemia and in 18% to 28% of patients with Down syndrome–associated ALL. In the case of Down syndrome–associated ALL, the mutations affect a highly conserved arginine residue (R683) in the pseudokinase domain of JAK2 and have been demonstrated to induce JAK/STAT activation and promote growth factor independence in BaF3 cells and response to JAK inhibitors.