21 Targeted Therapies for Gliomas
Since the publication of the previous edition of this book in 2008, our understanding of glioma tumor biology and associated molecular pathways and cellular alterations continues to progress rapidly. A more comprehensive understanding of the molecular basis of glioma initiation, progression, and treatment failure is emerging.1 The Cancer Genome Atlas established a comprehensive catalog of genomic alterations driving gliomagenesis, and identified several distinct molecular sub-types of glioblastoma that share common genetic mutations, methylation profiles, and molecular pathway activation. However, this growing insight into the molecular biology of glioma has not yet translated into effective therapies.2 Ongoing and future studies will evaluate specific inhibitors of these molecular targets, moving the field toward the development of personalized cancer therapy by which tumors with specific molecular alterations will be treated with agents inhibiting these activated pathways. These approaches will complement other strategies, such as targeting angiogenesis, glioma stem-like cells, and the immune response, with the ultimate goal of improving the outcome of patients with malignant gliomas.
 The Rationale for Targeted Therapy
The Rationale for Targeted Therapy
Several molecular alterations are thought to be important drivers of glioma tumorigenesis, growth, and treatment resistance, and provide the basis of the development of targeted therapies.3 Pharmacological inhibitors and modulators of these alterations have been tested in clinical trials, and further trials are ongoing. Most of these targeted therapies have been directed at receptor tyrosine kinases and their intracellular signaling pathways, identified preclinically as important for supporting tumor growth, proliferation, and angiogenesis.1 Despite these strategies, little has been accomplished in prolonging patient survival. Challenges include the genetic heterogeneity, inherent genetic instability, and rapid development of resistance pathways of gliomas, which make single-agent targeted therapies ineffective.1 Strategies including combination-agent targeted therapies and targeting multiple pathways are under design and evaluation, although treatment-related toxicity is a dose-limiting concern.1
Pitfall
• Combination therapy consisting of multiple targeted agents can be associated with markedly increased toxicity because of coexpression of multiple targets in normal cells.
Pearl
• Combination therapy consisting of targeted small-molecule inhibitors and cytotoxic therapy may provide a therapeutic advantage.
 Subclassification of Glioblastoma and Implications for Treatment
Subclassification of Glioblastoma and Implications for Treatment
Studies of gene expression and DNA methylation have identified multiple glioma subtypes, with some studies identifying an association between glioma subtype and survival.4–6 The Cancer Genome Atlas (TCGA) analysis confirmed three subtypes of high-grade glioma—proneural, mesenchymal, and proliferative—and identified the neural and classic subtypes.4–6 The proneural and mesenchymal subtypes are consistently identified and are the best characterized. The proneural sub-type is more frequently identified in younger patients, and is associated with platelet-derived growth factor receptor (PDGFR) amplification and isocitrate dehydrogenase (IDH) 1 mutation.5 The mesenchymal subtype is associated with neurofibromatosis type 1 (NF1) gene loss or mutation, and mesenchymal markers, such as MET.5 The neural subtype is associated with expression of neuronal markers,5 while the classic subtype is associated with phosphatase and tensin homolog (PTEN) loss, a distinct lack of TP53 mutation, and a high level of epidermal growth factor receptor (EGFR) alterations, found in over 95% of samples with this subtype.1,5 Grade II and III gliomas are predominantly of the proneural subtype, whereas glioblastomas can be of any subtype.5 Although one series suggested patients with proneural glioblastoma survive longer than those with mesenchymal glioblastoma, another study found no difference.5 Neural and classic subtypes are not clearly associated with different survival outcomes.1 Aggressive treatment with radiation with concurrent and adjuvant temozolomide chemotherapy significantly improved survival in classic and mesenchymal subtypes. A statistically nonsignificant association is observed in the neural subtype, but no significant effect on survival was observed in the proneural subtype.5 This subclassification of glioblastoma provides a foundation for a better molecular understanding of gliomagenesis and glioma signaling pathways and may be important for enriching clinical trials with patients screened for participation based on tumor molecular profile.1,5
 Critical Pathways in Gliomagenesis
Critical Pathways in Gliomagenesis
The mutation spectrum of glioblastoma, as described by TCGA, reveals that the majority of glioblastoma have aberrations in the TP53 pathway, RB pathway, or receptor tyrosine kinase (RTK) with downstream signaling through the Ras or phosphatidylinositol 3-kinase (PI3K) pathways.3,5 The majority of tumor samples analyzed had alterations in all three pathways, suggesting that dysregulation of these three pathways is key to gliomagenesis,7 although other pathways may also play critical roles3 (Fig. 21.1). In the TCGA cohort, inactivation of the p53 pathway occurred, for instance, in the form of ARF deletions, MDM amplifications, and mutations of p53 itself.7 Dysregulation of the RB pathway most commonly occurred with deletion of the CDKN2A/CDKN2B locus on chromosome 9p21, followed by CDK4 locus amplification.7 In the RTK/PI3K pathway, PTEN deletions and mutations were frequent, as were aberrations in EGFR, ERBB2, PDGFRA, and MET.7 Clearly, molecular heterogeneity and redundancy of molecular pathway activation will require careful selection of tumors for treatment with agents targeting the main drivers of tumor growth.
 Tumor Biomarkers
Tumor Biomarkers
Identifying and validating biomarkers are critical components of the development and evaluation of targeted therapies. Insight into glioma signaling pathways has helped uncover biomarkers that predict response to treatment and outcome in patients with glioma.3 Prognostic markers include O-6-methylguanine DNA-methyltransferase (MGMT) promoter methylation (epigenetic marker in glioblastoma, correlating with better prognosis), chromosomes 1p and 19q co-deletion (associated with better prognosis with treatment with radiotherapy plus chemotherapy), and IDH1 and IDH2 gene mutations (indicating better prognosis in gliomas). MGMT methylation may be predictive of response to alkylating chemotherapies, although this may be related to its strong prognostic effect. However, a lack of meaningful response with this potential predictive biomarker may relate to tumor heterogeneity of signaling pathway activation at a regional, subpopulation, or individual cell level.3 The somewhat recent discovery of IDH1 and IDH2 mutations in glioma enables a reproducible prognostic biomarker as well as a therapeutic target.1 IDH mutations have a significant positive statistical association with overall survival, and in low-grade gliomas, anaplastic gliomas, and glioblastomas, IDH mutation status appears to be the most important prognostic factor.1,8
Proneural glioblastoma with IDH mutations seem to segregate almost exclusively into a distinct CpG island hypermethylator phenotype (G-CIMP).1,9 This G-CIMP signature may be in fact the most robust prognostic indicator in glioblastoma, although the complexity and cost of determining G-CIMP status currently limit its utility as a routine prognostic test.1 There appears to be an almost complete overlap between G-CIMP status and IDH1/2 mutation status, which likely relates to the role of IDH mutations on the accumulation of epigenetic events leading to the hypermethylation phenotype.10 Although there is currently no alternative treatment for patients with a favorable or unfavorable molecular profile, incorporating these biomarkers is critical for patient stratification in clinical trials. Prospective identification of targets and genetic profiles of tumor tissue from patients who respond to specific treatments are now being introduced into clinical trials.
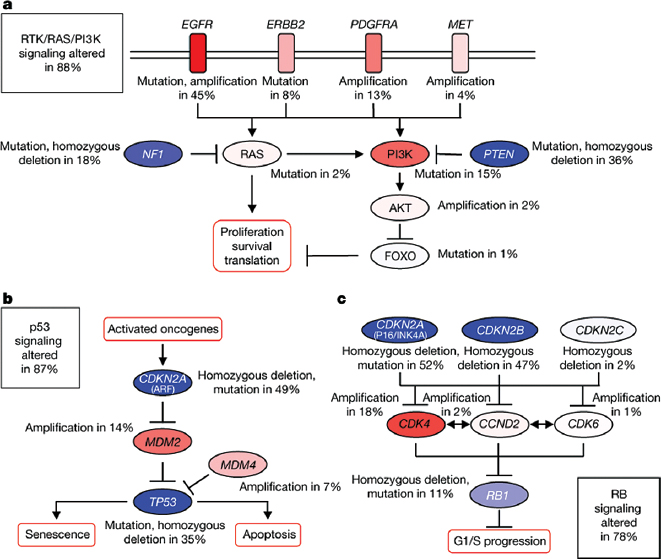
Fig. 21.1a–c Frequent genetic alterations in three critical signaling pathways. Primary sequence alterations and significant copy number changes for components of the (a) RTK/RAS/PI3K, (b) p53, and (c) RB signaling pathways are shown. Red indicates activating genetic alterations. Blue indicates inactivating alterations. The nature of the alteration and the percentage of affected tumors affected are indicated. Blue boxes contain the final percentages of glioblastoma with alterations in at least one known component gene of the designated pathway. (From Nature Publishing Group. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. The Cancer Genome Atlas Research Network, Fig. 5. Nature 2008;455:1061–1068. Reprinted with permission.)
 The Ideal Target
The Ideal Target
Ideally a target has all of the following characteristics: it is highly expressed on cancer cells; it is specific to tumor cells; it is causally related to tumor cell growth, proliferation, or invasion (tumor phenotype); and it is involved in tumor initiation (gliomagenesis). Gliomas have a multitude of genetic aberrations, and thus the most critical oncogenic target(s) and pathways that must be interrupted for cell death need to be identified. In addition, because tumors utilize multiple cellular pathways in a redundant manner, therapies targeted to a single molecule or pathway are likely to be ineffective.
Special Consideration
• Ideally, the target must be highly expressed in the tumor, the target must be active, and this activity must contribute to tumor growth, proliferation, and survival.
 Molecular Targets in Gliomas
Molecular Targets in Gliomas
As described above, specific genetic alterations identified in gliomas regulate several key pathways controlling cell growth, proliferation, invasion, and resistance to cell death. These highly complex processes involve signaling between multiple interrelated pathways. Key regulators of gliomagenesis include growth factor and growth factor receptors and their downstream effectors (i.e., molecules that mediate signal transduction and contribute to the transformed phenotype). Redundant and overlapping signaling pathways may explain, in part, the extreme resistance of these tumors to treatment. That is, attempts to inhibit a specific pathway within a tumor may result in the activation of a compensatory pathway or induce other cellular mechanisms that ultimately enable the tumor cells to survive. Nonetheless, some of the genetic alterations important in signal transduction pathways may be amenable to targeted therapy. Additionally, certain tumor suppressor genes such as PTEN and TP53 may also be important in determining tumor responsiveness to a specific molecular therapy, even though they may not represent a target for therapy.
Pitfall
• Single-agent therapy aimed at one target is unlikely to be effective in the treatment of gliomas because these tumors contain highly heterogeneous cell populations with multiple compensatory growth pathways.
Special Consideration
• The oncologist must have a working knowledge of glioma biology in order to understand the distribution and expression of the target in tumor and normal tissues and thus to anticipate the potential toxicities of molecular targeted therapies.
 Specific Therapeutic Targets and Relevant Clinical Trials
Specific Therapeutic Targets and Relevant Clinical Trials
Cell Surface Growth Factor Receptors
Several growth factor receptors frequently overexpressed in gliomas are oncogenes believed to “drive” glioma formation and progression, and therefore are major targets of drug development.11,12 These receptors, which predominantly act through receptor tyrosine kinases, are located on the cell surface, where they interact with effector molecules, second messengers, and other intracellular signaling pathway mediators, including the Ras/Raf/mitogen-activated protein kinase (MAPK) and PI3K pathways.12 These secondary pathways then activate additional downstream effectors, ultimately leading to enhanced cell survival, proliferation, migration and invasion, evasion of apoptosis, angiogenesis, and resistance to treatment.12 Growth factor pathways involved in glioma formation and progression include those mediated by EGFR, PDGFR, vascular endothelial growth factor (VEGF), transforming growth factor receptor (TGFR)-α and -β, and fibroblast growth factor receptor (FGFR)11 (Fig. 21.2). Glioma cells are also known to secrete growth factors for these receptors, which are overexpressed on the cell surface, thus establishing autocrine and paracrine growth stimulatory loops. The dominant role of these growth factors and their overexpression on glial tumors has made them central in the search for effective targeted therapeutic agents.
Epidermal Growth Factor Receptor
Epidermal growth factor receptor, a receptor tyrosine kinase that plays a fundamental role in the growth and transformation of primary glioblastoma, is a prime target for therapy.13 Indeed, by far the most frequent oncogenic alteration in gliomas is the overexpression, amplification, or mutation of EGFR, identified in approximately 60% of primary glioblastoma.13,14 EGFR expression has been associated with increased tumor cell proliferation, increased cellular migration and invasion, and increased resistance to chemotherapy. Aberrant EGFR signaling is less common in low-grade glioma and secondary glioblastoma that arise from lower grade tumors. Glioblastoma with the amplified EGFR gene are enriched in the classic subtype and are frequently associated with deletion of the CDKN2A gene locus.13

The EGFR pathway can be activated through multiple mechanisms, and initiates signal transduction through major pathways such as the MAPK cascade and PI3K/Akt13 to promote cell proliferation and survival. Some glioblastoma express EGFR and can also secrete the ligands EGF and TGF-α, thereby establishing autocrine and paracrine loops that effect the constitutive activation of the receptor and its downstream pathways. The most common EGFR mutation in glioblastoma is the intragenic deletion of exons 2 to 7, resulting in loss of the extracellular ligand-binding domain and constitutive activation of the receptor. This mutant receptor, referred to as EGFR vIII, is present in approximately 40% of tumors with EGFR amplification.11,14 EGFR vIII may be an independent prognostic factor for poor survival, as excessive activation of EGFR may provide cells with a growth advantage.11
• The expression of a target may not predict the responsiveness of a tumor to target inhibition because of differences in patient populations, genetic heterogeneity within the tumor, and the relative inefficiency of the small molecule in inhibiting the target.
There are several reasons why EGFR and its downstream pathways are ideal targets for therapy. First, as noted above, EGFR is highly expressed in gliomas. Second, EGFR overexpression correlates with poor patient prognosis (decreased survival time and shorter time to progression). Third, EGFR plays no critical physiological role in healthy adults.
The first-generation single-agent small-molecule tyrosine kinase inhibitors (TKIs) used in clinical trials targeting EGFR in glioblastoma were gefitinib (ZD1839; Iressa) and erlotinib (OSI-779; Tarceva). Although the drugs were well tolerated with mild side-effect profiles, no treatment benefit was found.13 It should be noted that these trials were offered to unselected patients with glioblastoma and the pharmacokinetics of these EGFR inhibitors were in some cases likely altered by the concurrent use of enzyme-inducing antiepileptic drugs.13 Independent trials did not confirm a benefit in patients with EGFR vIII expression and PTEN loss,15 as was previously reported.16 Failure of the first-generation EGFR inhibitors in glioma clinical trials may have been related to inadequate drug levels in the tumor, the inability to turn off the receptor for a sufficient period of time, and the rapid activation of resistance pathways. Future trials will attempt to minimize the confounding impact of these important factors when evaluating newer agents.
Limited efficacy was observed with cetuximab (Erbitux, ImClone Systems), a monoclonal antibody against EGFR and lapatinib (GW572016, GlaxoSmithKline USA, Philadelphia, PA), a TKI targeting HER1 and HER2 (Tan). Cetuximab has limited penetration across the blood–brain barrier, which may have been responsible for its limited clinical activity.11 Preclinical studies have identified lapatinib as an irreversible inhibitor of EGFR, which may confer greater therapeutic benefit over previous TKIs such as erlotinib.16 Ongoing studies with lapatinib will determine if intermittent high-dose (pulsed-dosing) strategies can improve the efficacy of EGFR inhibition. Next-generation TKIs, such as afatinib, dacomitinib, and nimotuzumab, which have reversible EGFR inhibition, are under development and testing.3
Combining erlotinib with other therapies, including radiation therapy, cytotoxic chemotherapy, and other smallmolecule therapies, failed to improve its efficacy. A phase 2 trial of EGFR and mammalian target of rapamycin (mTOR) inhibition was ineffective, but patients were not stratified for a specific molecular signature. Several studies have shown a response typically in tumors with EGFR amplification and low levels of Akt or expression of the EGFR vIII variant and preservation of PTEN, suppressing constitutive Akt activation, providing rationale for stratifying patients.
The basic principle of targeted therapy is that tumors expressing the target will respond to a particular drug designed to inhibit that target. However, although the concept is simple, realizing this clinically has not proved to be straightforward. Molecular testing of glioblastoma tissue of patients under treatment has established drug concentrations in the tissue, and has helped further our understanding of the effects on pathway signaling.13 However, few or no responders in these clinical trials of EGFR inhibitors suggests that there is not a “signature” that will inform efficacy. Vaccination approaches targeting the EGFR vIII mutant with a tumor-specific antigen are ongoing in randomized phase 3 clinical trials.13
Platelet-Derived Growth Factor Receptor
Similar to EGFR, the stimulation of PDGFR activates downstream signaling cascades involving Ras/Raf/MAPK and PI3K/Akt pathways, and is involved in promoting cell survival, proliferation, invasion, and resistance to apoptosis. In addition, similar to other receptor tyrosine kinases, both the ligand (PDGF) and its receptor (PDGFR
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


