Fetal globin (gamma globin; HBG) is normally expressed during fetal life and prevents the clinical manifestations of beta hemoglobinopathies before birth. HBG genes are normally integrated in hematopoietic stem cells in all humans, and are at least partially amenable to reactivation. Inducing expression of fetal globin (HBG) gene expression to 60% to 70% of alpha globin synthesis produces a β-thalassemia trait phenotype, and reduces anemia. Tailoring combinations of therapeutics to patient subsets characterized for quantitative trait loci which modulate basal fetal hemoglobin and erythroid cell survival should provide effective amelioration of clinical symptoms in β-thalassemia and sickle cell disease.
Key points
- •
Fetal globin reduces clinical events initiated by sickle hemoglobin polymerization, because it cannot participate in the process.
- •
Fetal globin chains reduce excess alpha globin and globin chain imbalance in β-thalassemia, improving total hemoglobin levels.
- •
Target levels of fetal globin that reduce clinical severity in sickle cell disease and β-thalassemia are established from natural mutations, modifiers, and from treatment trials.
- •
Genetic modifiers related to the beta globin locus and/or to erythroid cell stress signaling and survival influence responses to different therapeutics.
- •
Combinations of therapeutics with differing molecular mechanisms and that promote erythroid survival offer new opportunities for personalized, highly active treatment.
Introduction
β-Thalassemia syndromes are common monogenic disorders worldwide, characterized by molecular mutations that cause deficiency of the beta globin chain of adult hemoglobin (HbA; α 2 β 2 ), and an excess of unmatched alpha globin chains. Excess alpha globin damages the red blood cell membrane and causes apoptosis of developing erythroblasts and intramedullary hemolysis. Clinical observations and previous trials of fetal globin inducers have shown that patients with β-thalassemia benefit from natural persistence of, or pharmacologic induction of, another type of globin that is normally suppressed before birth and in infancy: fetal hemoglobin (HbF; HBG, gamma globin). Patients with higher gamma globin levels than their counterparts with the same mutations often do not require transfusions as regularly or as early in life as patients with lower levels of gamma globin. Inheritance of a single modifying trait that increases HbF, such as a single nucleotide polymorphism (SNP) in BCL11A, without any other genetic difference, can produce higher total hemoglobin up to 1 g/dL. The impact of HbF is particularly notable in infants with sickle cell disease (SCD), who survive in utero in a highly hypoxic environment that would produce completely sickled cells with no oxygen delivery without the presence of HbF in every red blood cell. Other natural models of the benefit of increased HbF include sickle cell populations with milder disease and greater than or equal to 20% HbF in Saudi Arabia and in India (the Arabian Indian haplotype), whereas 30% HbF in S-HPFH produces a benign condition. The National Institutes of Health Natural History Study and multiple studies of hydroxyurea (HU) showed the highly significant ameliorating effects of HbF at levels greater than 8.6% or 0.5 g/dL. In addition, treatment trials, such as those conducted with arginine butyrate, have increased HbF from a mean of 7% to 21% and reduced hospital days by 3-fold. Inducing gamma globin expression by even small increments is recognized as a therapeutic avenue that should be amenable to broad application, because the gamma globin genes are universally present and normally integrated in hematopoietic stem cells. Although a single chemotherapeutic drug, hydroxyurea, is commercially available and has had variable effects, several important principles for this approach have been defined in trials of prior generations of therapeutics. The recent discovery of new therapeutic candidates now offers a renaissance for this approach.
Introduction
β-Thalassemia syndromes are common monogenic disorders worldwide, characterized by molecular mutations that cause deficiency of the beta globin chain of adult hemoglobin (HbA; α 2 β 2 ), and an excess of unmatched alpha globin chains. Excess alpha globin damages the red blood cell membrane and causes apoptosis of developing erythroblasts and intramedullary hemolysis. Clinical observations and previous trials of fetal globin inducers have shown that patients with β-thalassemia benefit from natural persistence of, or pharmacologic induction of, another type of globin that is normally suppressed before birth and in infancy: fetal hemoglobin (HbF; HBG, gamma globin). Patients with higher gamma globin levels than their counterparts with the same mutations often do not require transfusions as regularly or as early in life as patients with lower levels of gamma globin. Inheritance of a single modifying trait that increases HbF, such as a single nucleotide polymorphism (SNP) in BCL11A, without any other genetic difference, can produce higher total hemoglobin up to 1 g/dL. The impact of HbF is particularly notable in infants with sickle cell disease (SCD), who survive in utero in a highly hypoxic environment that would produce completely sickled cells with no oxygen delivery without the presence of HbF in every red blood cell. Other natural models of the benefit of increased HbF include sickle cell populations with milder disease and greater than or equal to 20% HbF in Saudi Arabia and in India (the Arabian Indian haplotype), whereas 30% HbF in S-HPFH produces a benign condition. The National Institutes of Health Natural History Study and multiple studies of hydroxyurea (HU) showed the highly significant ameliorating effects of HbF at levels greater than 8.6% or 0.5 g/dL. In addition, treatment trials, such as those conducted with arginine butyrate, have increased HbF from a mean of 7% to 21% and reduced hospital days by 3-fold. Inducing gamma globin expression by even small increments is recognized as a therapeutic avenue that should be amenable to broad application, because the gamma globin genes are universally present and normally integrated in hematopoietic stem cells. Although a single chemotherapeutic drug, hydroxyurea, is commercially available and has had variable effects, several important principles for this approach have been defined in trials of prior generations of therapeutics. The recent discovery of new therapeutic candidates now offers a renaissance for this approach.
Experience in trials of prior generation HbF inducers
Table 1 lists several therapeutics that induce HbF and are being clinically investigated. Proof of principle of HbF induction was shown in previous clinical trials of several drugs in which pharmacologic reactivation of gamma globin expression reduced anemia and even eliminated transfusion requirements in patients with β-thalassemia. HbF induction has been accomplished with chemotherapeutic agents, particularly 5-azacytidine, and 5-aza-2-deoxycytidine (decitabine), and with short-chain fatty acids (SCFAs), such as arginine butyrate (AB) and sodium phenylbutyrate. 5-Azacytidine and decitabine have shown high potency with responses in 12 of 13 patients in one study, including adult patients with SCD who do not respond to hydroxyurea. Cellular abnormalities were all reduced after HbF levels were increased.
| Therapeutic Class | Therapeutic Name | Current Development Status/Activity |
|---|---|---|
| Demethylating agents | 5-Azacytadine, decitabine | Potent HbF inducer; IV use only; myelosupressive, mutagenic Low-dose metronomic regimen promotes erythroid differentiation, active in phase 2 trials |
| Decitabine + THU | THU inhibits metabolizing enzyme, allows oral use Active in baboons; in phase I–II SCD dosing trial in progress | |
| Ribonucleotide reductase inhibitor | Hydroxyurea (Droxia) | Activity through transient cytostasis; sole drug approved for SCD, oral High activity in young children; beneficial in 50% of adults; increases mean HbF by 2%–3%, reduces clinical events in SCD; improves erythropoiesis, increases mean total Hgb in HbE β-thalassemia by 0.6 g/dL. Clinical benefit, improved SCD survival with HbF >0.5 g/dL |
| HDAC inhibitors | Vorinostat | Oral; in phase1/2 trial in SCD |
| Panobinostat | Oral, active in transgenic sickle mice | |
| HDAC1/2 inhibitors MS-275 | In vitro activity, in preclinical development. Active in vitro and in vivo in baboons, in phase 3 cancer trial; suppresses BCL11A | |
| Short-chain fatty acid derivatives | Arginine butyrate | Intermittent dosing increased HbF 3-fold in SCD, and total Hgb >3 g/dL in β-thalassemia; low bioavailability (IV injection) HDAC inhibitor, suppresses BCL11A , displaces HDAC3 from gamma promoter |
| Sodium phenylbutyrate | Oral, low bioavailability, requires large doses, increases HbF in SCD and total Hgb in 50% of patients with thalassemia | |
| Sodium 2,2-dimethylbutyrate | Oral derivative, displaces HDAC2 from promoter; promotes erythroid survival through Bcl-xL survival protein; long half-life, active in patients with SCD and β-thalassemia, in phase 2 trials | |
| Thalidomide derivatives | Pomalidomide | In vitro and in vivo transgenic mice activity, activity observed in phase 1–2 trials in SCD |
| LSD-1 inhibitors | Tranylcypromine | Inhibits corepressor LSD-1, which binds TR2/TR4 nuclear receptors in the gamma globin promoter to silence expression; active in β-YAC mice; use in psychiatric disorders |
| Benserazide | Displaces HDAC3, LSD-1 from promoter, active in SCD/thalassemia progenitors, baboons, transgenic mice; long-term clinical use as PK enhancer for amino acids | |
| Stress signaling enhancer | Salubrinal | Inhibits dephosphorylation of, and increases eIF2αP, with enhanced stress signaling and thalassemic erythroid survival, promotes translation of HbF |
A therapeutic that is not cytotoxic is preferable for a long-term therapy in β-thalassemia, because with cumulative dosing with hydroxyurea total hemoglobin (Hgb) levels increase, usually by less than 1 g/dL but also tend to decline over time. The first-generation SCFAs had limitations of rapid metabolism and high dose requirements; AB and phenylbutyrate are also global histone deacetylase (HDAC) inhibitors that inhibit erythropoiesis through cell cycle arrest. Erythropoiesis-stimulating agents are beneficial, but require parenteral administration and are too costly for lifelong therapy. Nevertheless, these three classes of therapeutics reduced anemia and rendered some patients with thalassemia transfusion independent. Therapeutics that require lower doses and oral administration would allow broader application, particularly in regions where thalassemia is common globally and transfusions carry particularly high risks of infection.
Several observations in the earlier trials were informative regarding magnitude of responses, and patterns of response, in patients with differing β-thalassemia mutations. Several inducers (5-azacytidine, phenylbutyrate, AB, and erythropoietin [EPO]) produced significant hematologic responses with increases in total hemoglobin of 2 to 5 g/dL or more greater than baseline. Although the clinical trials have been small, patients with diverse thalassemia syndromes had significant responses, including transfusion independence. Collins and colleagues found that sodium phenylbutyrate increased total Hgb by 2 g/dL greater than baseline, and that responses occurred more frequently in patients with EPO levels greater than 160 mU/mL. Increases in total Hgb levels of 1 to 5 g/dL greater than baseline were achieved when these agents were administered for at least 3 to 6 months. This finding is remarkable, because thalassemic cells survive for only a few days, compared with the normal red cell survival of 120 days.
Of the chemotherapeutic agents, hydroxyurea (HU) treatment has increased total Hgb by 0.6 to 1.0 g/dL in patients with HbE/β-thalassemia, and, although the effect was not as great in magnitude, it significantly reduced hemolysis, which is a cause of many complications. Hajjar and Pearson reported that gamma globin increased rapidly with HU treatment, with a 6-week treatment time frame required for a peak response, but that it was followed by a decline in total Hgb, suggesting cellular growth inhibition. 5-Azacytidine increased total Hgb levels by an average of 2.5 g/dL (range 1–4 g/dL), even in end-stage patients with life-threatening severe anemia.
Of the SCFAs and HDAC inhibitors, AB, administered first frequently, 4 to 5 days/wk, and then intermittently, twice per month, increased total Hgb levels by 1 to 5 g/dL (mean 2.9 g/dL) when administered for 3 to 6 months. AB treatment rendered patients transfusion independent for several years with home therapy, given 4 nights every other week to avoid the (reversible) antiproliferative effects common to HDAC inhibitors. In a trial using pulse, or intermittent, AB, HbF increased by a mean of 3-fold in 9 of 11 patients, increasing from 7% to 21%, and hospital days were reduced from 80/y to 20/y. AB has been safe in long-term use, with no significant butyrate-related adverse events in more than 16 patient-years of home administration provided by parents. EPO preparations increased Hgb levels by 1 to 3 g/dL more than baseline in patients with thalassemia intermedia, and decreased transfusion requirements in thalassemia major. HbF did not increase with EPO or darbopoietin therapy, so that only thalassemic red blood cell production increased, rather than red cells corrected for globin chain imbalance as occurs with the HbF inducers. However, EPO trials highlight the importance of maintenance of healthy erythroid cells for fetal globin to be induced by any pharmacologic or biologic treatment. These trials all show proof of principle of the usefulness of therapeutic induction of gamma globin with or without enhancement of erythropoiesis in patients with β-thalassemia and SCD.
Molecular targets: HBG globin transcription and the fetal globin program
The temporal and tissue-specific expression of the betalike globin genes, including ζ, A γ, G γ, δ, and β, are controlled at multiple levels. Within the beta globin cluster, there are cis -acting promoter elements for each gene and a distal trans -acting element designated the locus control region (LCR). Looping of the LCR in close proximity to the individual cis -promoter elements and competition between the globin genes for interaction with the LCR regulates their developmentally regulated expression. Mice transgenic for constructs containing the beta and gamma globin genes and their cis -acting elements, together with the LCR, show appropriate stage-specific and tissue-specific expression of fetal and adult globin.
There is a broad range of basal HbF globin levels among individuals, defined by their inherited fetal globin program. SNPs and mutations/deletions within the beta globin cluster account for a portion of this variation. Genetic loci beyond the beta globin cluster, which regulate the fetal globin program, have recently been identified by genome-wide association studies (GWAS) and subsequently validated. Two of these elements, the HBS1L-MYB intergenic interval on chromosome 6 and BCL11A on chromosome 2, seem to be responsible for an estimated 15% to 20% respectively of individual variation in HbF globin levels. Since their discovery, correlative studies have shown that SNPs within these loci that result in increased levels of HbF ameliorate the severity of SCD and β-thalassemia. Certain mutations within the KLF1 transcription factor also result in persistent high-level expression of gamma globin after birth.
HBS1L-MYB Intergenic Interval
c-Myb is a transcription factor, initially identified as a proto-oncogene in leukemogenic retroviruses, which is required for definitive hematopoiesis. The activity/level of Myb is reciprocally related to the basal level of fetal globin expression.
KLF-1 (EKLF)
KLF-1 is a transcription factor that binds to a CACCC element in the beta promoter (and a similar element in the gamma A and gamma G promoters, albeit with lesser affinity) and drives high-level adult-stage beta globin expression. Repression of KLF-1 activity reduces beta globin transcription and reciprocally enhances gamma globin expression, reversing its developmental silencing.
BCL11A
BCL11A was identified as a major regulator of basal fetal expression patterns in GWAS and was later shown to be a transcriptional repressor that is required for the maintenance of silencing of fetal or embryonic globin expression in human and murine erythroid cells, respectively. BCL11A interacts with multiple erythroid transcription factors and with the NuRD nucleosome remodeling a deacetylase complex, promoting physical interactions between the LCR and the beta globin promoter, enhancing beta globin transcription, and reciprocally suppressing gamma globin. These genetic modifiers of the fetal globin program also show interactive regulation of each other. c-Myb influences KLF-1 expression. The KLF-1 protein, in addition to directly activating beta globin transcription, also activates BCL11A expression, which also directly induces beta globin expression.
Targeted gamma globin activation through the CACCC element
Understanding fetal globin gene regulation by identifying various cis -acting elements and their interacting trans -acting factors has been a goal for many years to develop therapeutic targets. A key element regulating the betalike globin genes is the CACCC box, located in the proximal promoter ( Fig. 1 ). Naturally occurring mutations in the beta-CACCC box cause β-thalassemia and deletion of the gamma-CACCC box significantly attenuates gamma globin promoter activity. Transcription factors belonging to the Sp1/Krüppel-like factor (KLF) family recognize CACCC/GC boxes to regulate gene transcription. The founding member KLF-1 (EKLF) is an erythroid-specific factor and positive regulator of the beta globin gene. KLF-1 is critical for hemoglobin switching, and targeting this factor has not been achieved for effective therapy for the hemoglobinopathies. Recent insights into repressive factors such as KLF-1 that silence gamma globin expression during development offer new approaches to reactivate HbF expression. Knockdown of the repressor factor BCL11A , known to be activated by KLF-1, increases HbF in cultured erythroblasts and ameliorates the SCD phenotype in transgenic mice. Transcription factors involved in gamma globin activation earlier in development remain to be discovered.
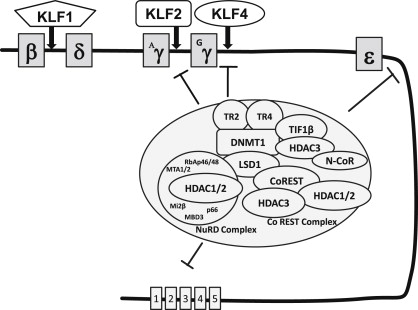
Other KLF family members have been investigated for their roles as gamma globin regulators. Previous reports showed that KLF-13 (FKLF-2), associates with creb binding protein (CBP)/p300 to activate gamma globin transcription, although FKLF-2 did not enhance the endogenous gene. A subsequent study in K562 cells showed that KLF-2, KLF-5, and KLF-13 positively regulate and KLF-8 negatively regulates the gamma globin gene through the CACCC element. Furthermore, an essential role for KLF-4 in primitive erythropoiesis was shown in zebrafish. Pace and colleagues recently defined KLF-4 as a positive regulator of gamma globin expression in human primary erythroid cells through KLF-4 binding to the CACCC box; moreover, CBP inhibits the ability of KLF-4 to activate the gamma globin promoter. These studies define novel molecular mechanisms by which the Sp1/KLF family of factors might be targeted for therapeutic gamma globin gene activation.
Therapeutic approaches directed to increasing gamma globin transcription
The identification of transcription factors and other genetic modifiers that influence the fetal globin program in a cell suggests the possibility of targeting these components directly as a method to enhance HbF expression in the beta hemoglobinopathies. These modifiers were validated as targets using genetic methodology, including knock-outs and small hairpin RNA, but these approaches do not yet have clinical application. Instead, less direct approaches, using small molecules that affect more readily drugable components of transcriptional activator or repressor complexes, including epigenetic modifiers, have been the first to be tested clinically. Table 1 shows several drug classes that are, or have been, clinically studied.
Demethylation of the Silenced Gamma Globin Genes
The first such approach used 5-azacytadine, a DNA demethylating agent. It had been observed that the silenced gamma globin genes in the adult were heavily cytosine methylated, as were other genes known to be silenced after embryonic or fetal life (although it was later shown that the methylation occurred long after the gamma globin genes were transcriptionally silenced). 5-Azacytidine is a potent inhibitor of the enzyme responsible for maintaining this methylation, DNA N-methyl transferase (DNMT1). The hypothesis that the gamma globin genes could be reactivated by this DNA demethylating agent was validated in primate studies and small clinical studies in which demethylation of the fetal globin genes and their coincident reactivation were shown. Because the demethylating activity of 5-azacytidine could not be separated from its potent cytotoxicity, interpretation of these studies was confounded as to whether the mechanism of reinduction was via direct demethylation or indirectly via stress erythropoiesis. More recently, support for this approach has been generated using decitabine, a metabolite of 5-azacytidine that retains its potent DNMT1 inhibitory activity, but is far less cytotoxic, and more effective when used in a low dose on a regular, but intermittent (metronomic) regimen. Decitabine has been shown in clinical studies of SCD and β-thalassemia to be a potent inducer of HbF, with activity even in severely affected adult patients who do not respond to hydroxyurea. Transcriptional repression of the fetal globin gene is accomplished in large part by site-specific binding of a DNA-binding protein and subsequent recruitment of several different types of corepressor proteins that contribute to gene silencing. For example, the nuclear receptors TR2 and TR4 (TR2/TR4) bind to direct repeat elements in human embryonic and fetal globin gene promoters and play a critical role in the silencing of these genes by recruiting DNMT1, and the NuRD and LSD-1/CoREST repressor complexes and histone deacetylase 3 (HDAC3), which then effect coordinated epigenetic chromatin modifications. Deacetylation of histones and the consequent formation of nucleosomes and resulting compaction of chromatin is a primary epigenetic mechanism of gene silencing. HDAC2 and HDAC3 have also been identified at the silenced gamma globin promoter by chromatin immunoprecipitation. Knockdown of HDAC3 by small interfering RNA is sufficient to induce transcription of the silenced gamma globin gene promoter in primary erythroid progenitors. In a similar way, knockdown of HDAC1 or HDAC2 has been shown to be sufficient to induce gamma globin gene expression in some in vitro systems and is now being studied in progenitors cultured from patients with hemoglobinopathies.
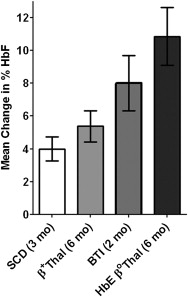
The first HDAC inhibitory agent tested clinically in the beta hemoglobinopathies was the short-chain fatty acid arginine butyrate (AB), which, when used in frequent and then intermittent dosing regimens, produced significant hematologic and clinical responses in 9 of 11 patients with SCD and long-term elimination of transfusion dependency in patients with β-thalassemia. Its activity partly led to current evaluation of several oral HDAC inhibitors. Pulse administration overcame the reversible growth inhibitory activity of AB and phenylbutyrate (SCFAs). Irreversible cytotoxicity of other HDAC inhibitors, including benzamides, cyclic tetrapeptides, and hydroxamates, may be more challenging for effective dosing, but are worthwhile to investigate. New generations of short-chain (aliphatic) fatty acids have been developed through pharmacophore modeling that lack HDAC inhibitory activity and do not inhibit erythroid progenitor cell proliferation. At least one such derivative, sodium 2,2-dimethylbutyrate (SDMB), has been studied in dose-ranging trials in both SCD and β-thalassemia, has induced HbF in both conditions to different degrees in various conditions ( Fig. 2 ), and is now in late phase II clinical trials in SCD.
Related posts:
 Ischemia-reperfusion Injury in Sickle Cell Anemia
Ischemia-reperfusion Injury in Sickle Cell Anemia
 Gene Therapy for Hemoglobinopathies
Gene Therapy for Hemoglobinopathies
 Alterations of the Arginine Metabolome in Sickle Cell Disease
Alterations of the Arginine Metabolome in Sickle Cell Disease
 Ischemia-reperfusion Injury in Sickle Cell Anemia
The Role of Adenosine Signaling in Sickle Cell Therapeutics
Ischemia-reperfusion Injury in Sickle Cell Anemia
The Role of Adenosine Signaling in Sickle Cell Therapeutics
 Alterations of the Arginine Metabolome in Sickle Cell Disease
Alterations of the Arginine Metabolome in Sickle Cell Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


