Ischemia-reperfusion (I/R) physiology, also called reperfusion injury, instigates vascular and tissue injury in human disease states. This review describes why sickle cell anemia should be conceptualized in this fashion and how I/R physiology explains the genesis of characteristic aspects of vascular pathobiology and clinical disease in sickle cell anemia. The nature of I/R and its relevance to sickle cell anemia are discussed, with an emphasis on the acute chest syndrome, endothelial dysfunction with aberrant vasoregulation, circle of Willis vasculopathy, and inflammatory pain. Viewing sickle disease from this perspective elucidates defining pathophysiology and identifies a host of novel potential therapeutic targets.
Key points
Disparate clinical features and vascular biological abnormalities characteristic of sickle cell anemia can be explained readily by viewing this disease as an example of ischemia-reperfusion (I/R) injury, which establishes a chronic, systemic inflammatory state.
- •
In sickle I/R, the triggering ischemia probably occurs in the microvasculature and in a dispersed, multifocal manner.
- •
Features of sickle I/R are influenced by the multitude of modulating factors present in its uniquely complex milieu.
- •
Acute chest syndrome may be an example of I/R-induced remote organ injury.
- •
Other sickle disease features explainable by the I/R character of sickle disease include endothelial dysfunction with aberrant vasoregulation, large vessel vasculopathy, and inflammatory pain.
Introduction
Ischemia-reperfusion (I/R) physiology, also called reperfusion injury, is a fundamental vascular pathobiological paradigm, which instigates vascular and tissue injury in a variety of human disease states. This review describes why sickle cell anemia should be conceptualized in this fashion and how I/R physiology explains the genesis of characteristic aspects of vascular pathobiology and clinical disease in sickle cell anemia. For clarity, the nature of I/R generally is presented first, followed by an exposition of the relevance of this information to sickle cell anemia. The clinical complications to be emphasized are the acute chest syndrome (ACS), endothelial dysfunction with aberrant vasoregulation, circle of Willis vasculopathy, and inflammatory pain. Viewing sickle disease from this perspective elucidates defining pathophysiology and identifies a host of novel potential therapeutic targets.
Introduction
Ischemia-reperfusion (I/R) physiology, also called reperfusion injury, is a fundamental vascular pathobiological paradigm, which instigates vascular and tissue injury in a variety of human disease states. This review describes why sickle cell anemia should be conceptualized in this fashion and how I/R physiology explains the genesis of characteristic aspects of vascular pathobiology and clinical disease in sickle cell anemia. For clarity, the nature of I/R generally is presented first, followed by an exposition of the relevance of this information to sickle cell anemia. The clinical complications to be emphasized are the acute chest syndrome (ACS), endothelial dysfunction with aberrant vasoregulation, circle of Willis vasculopathy, and inflammatory pain. Viewing sickle disease from this perspective elucidates defining pathophysiology and identifies a host of novel potential therapeutic targets.
I/R injury
The basic concept of I/R is that tissue injury resulting from vascular occlusion occurs in 2 distinct phases. The first is direct ischemia-induced tissue injury or death, determined primarily by severity and duration of blood supply interruption. If occlusion resolves, allowing reperfusion of the previously ischemic area, the accompanying resupply of oxygen triggers a second, inflammatory phase. The latter establishes systemic inflammation and its sequelae, and sometimes results in remote organ injury (ROI) or even multiorgan dysfunction syndrome (MODS). This complex aspect of biomedicine is described by 2 reviews covering different decades of research using experimental animal models of I/R. Additional citations are provided here when specific points require emphasis. This review emphasizes features that most clearly are specifically relevant to the sickle disease context.
Initiation of I/R
An occlusion causing ischemia triggers loss of adenosine triphosphate and reciprocal accumulation of hypoxanthine. This process is followed by cytosolic calcium accumulation, mitochondrial dysfunction, cell swelling, and cell death by inflammatory (necrosis) and noninflammatory (autophagy) mechanisms. In parallel, there is a dramatic change in xanthine dehydrogenase (XD), a widely distributed cellular enzyme that is particularly enriched in capillary endothelial cells, hepatocytes, and intestinal enterocytes. During hypoxia, XD is converted to its xanthine oxidase (XO) form, and significant amounts can be released into the blood space. Within minutes of restoration of reperfusion, XO begins using the accumulated hypoxanthine plus newly available oxygen to fuel superoxide radical generation. In turn, this process enables generation of other reactive oxygen species (ROS), events promoted by iron bioavailability. Thus, XO is understood to be largely responsible for the proximate burst of ROS that is believed to initiate the overall I/R process, resulting in I/R injury.
The Further Evolution of I/R
Subsequent events rapidly become complex, with recruitment of an alarming panoply of cellular, vascular, and parenchymal cell processes that follow from I/R initiation. Additional sources of superoxide radical become activated. Blood-borne XO becomes associated with endothelial cell surfaces, thereby establishing superoxide generation at the endothelial surface. Activity of endothelial nicotinamide adenine dinucleotide phosphate (NADPH) oxidase increases, and endothelial dysfunction ensues with uncoupled endothelial nitric oxide synthase (eNOS) (see later discussion). Leukocytes become activated and generate oxidant. Other superoxide sources can contribute as well. Effects of ROS are indirect (caused by peroxide-based disruption of thiol redox status), modulatory (via effects on cell signaling), and damaging. Oxidant stress causes widespread and varied oxidative and nitrosative damage, which can adversely affect virtually all biological molecules.
The ischemic area acquires multiple activated leukocyte types (polymorphonuclear neutrophils [PMN], monocytes, lymphocytes), dominated initially by PMN infiltration. Sometimes, the concordant endothelial and PMN activation leads to leukocyte-mediated microvascular plugging, which can even precipitate a no-reflow phenomenon, with local expansion of the original area of ischemic injury. Activation of platelets, endothelial cells, tissue resident macrophages, and mast cells initiates elaboration of a host of inflammatory substances that cause local inflammation and injury.
Of these biological mediators, tumor necrosis factor α (TNF-α) is most strongly implicated as a proximate promoter, its principal producers being blood monocytes, tissue resident macrophages, and perivascular mast cells. In turn, TNF-α induces chemokine production, further ROS production, and critical activation of nuclear factor κB (NFκB), the master trigger of inflammatory gene expression. These events occur explosively, because NFκB activation begins within minutes of onset of reperfusion.
Through these mechanisms, various additional inflammatory mediators appear, in particular: platelet activating factor (PAF), leukotriene B 4 (LTB 4 ), monocyte chemoattractant protein 1 (MCP-1), vascular endothelial growth factor (VEGF), and, in some models, complement component C5a. Toll-like receptor 2 (TLR2) and TLR4 signaling (in leukocytes, endothelial cells, parenchymal cells, mast cells), prompted by classic TLR ligands generated by cell damage, independently initiates inflammatory cytokine responses.
As part of this overall process, activation of mast cell plays a prominent amplification role. Their many activators include superoxide, PAF, and possibly IgE. By secretion or degranulation, mast cells release TNF-α, interleukin 6 (IL-6), and IL-1β, VEGF, LTB 4 , PAF, tryptase, histamine, and other agents. Activation of monocytes, tissue macrophages, mast cells, natural killer cells, and other lymphocyte subsets can exert injurious effects on tissues.
Despite the complexity of this spectrum of biological processes and mediators involved in I/R injury, some emerge as thematically dominant (ie, as master triggers and effectors: a proximate burst of superoxide generation, classically via XO activity; early provocation by TNF-α; mediation by endothelial NFκB activation; infiltration by PMN; amplification by mast cells; and probably attempted mitigation by heme oxygenase 1 [HO-1]).
Differential Organ Susceptibilities
Experimental animal studies show that different organs have differing susceptibilities to developing injury if local I/R occurs; these are in descending order of susceptibility: brain; myocardium, kidney, intestine; liver, skeletal muscle; lung. Direct injury from the initial ischemia develops relatively rapidly, but full development of additional local injury from the reperfusion phase can extend over several days. The pace and the severity threshold at which the former versus latter components develop can vary from organ to organ, thus explaining some of the differential susceptibilities.
The susceptibility of the brain derives from its multiple atypical features: the highest metabolic activity; its absolute metabolic requirement for glucose, despite low local storage; and its high polyunsaturated fatty acid content, despite lower levels of antioxidant enzymes.
Conversely, the lung is uniquely resistant to local initiating ischemia because of its abluminal high oxygen availability. When it does occur, I/R within the lung is complicated, because oxygen tensions of blood (bronchial artery) and tissue (alveolus) can vary independently.
Systemic Implications
A key theme of I/R is that it unleashes a systemic inflammatory response, a process potentially so robust as to generate MODS. On reperfusion, activated blood cells, inflammatory mediators, and other endothelial cell perturbing factors enter the circulation and travel to sites remote from the original occlusion. Activated endothelial cells support capture, adhesion, and transmigration of previously activated PMN. Activated platelets can deposit inflammatory chemokines on the endothelial surface.
Endothelial Dysfunction
During these events, endothelial cells attempt to integrate discordant signals into an appropriate response, but the inflammatory onslaught nudges them toward a maladaptive state. Increased activity of endogenous NADPH oxidase shifts oxidant impact away from normal mediation of homeostatic signaling and toward exertion of oxidant stress ; newly acquired endothelial surface XO contributes. This situation results in oxidative loss of tetrahydrobiopterin and perhaps of its rate limiting formative enzyme ; this process uncouples eNOS, causing it to generate superoxide rather than nitric oxide (NO).
This combination of enhanced oxidative stress plus diminished NO bioavailability is characteristic of endothelial dysfunction, a state that also includes increased vascular permeability and a shift toward prothrombotic and proinflammatory endothelial cell phenotypes. Endothelial cells themselves begin generating inflammation-promoting agents (eg, MCP-1, IL-1, and PAF). The biodeficiency of NO subverts its normal inhibitory effect on platelet activation, leukocyte adhesion, NFκB-mediated transcription, vasoconstriction, superoxide excess, and Weibel-Palade body release of von Willebrand factor and P-selectin. In addition, the inflammatory state triggers degradation of the endothelial glycocalyx, which normally provides husbandry of homeostatic endothelial functions. Thus, systemic endothelial dysfunction involving both conduit and microvascular vessels is a typical feature of I/R injury states.
ROI
As a truly hallmark feature of I/R, it can initiate injurious effects at sites remote from the site of initial ischemia: ROI or even MODS. In experimental models, such potentially catastrophic complications derive from I/R originating in heart, intestine, lung, kidney, muscle, and liver. Features of I/R-induced systemic inflammation that are particularly pathogenic in ROI include deposition of XO on endothelial surfaces and concurrent activation of leukocytes and endothelium.
The lung is the most commonly involved ROI target organ in both experimental and human disease I/R states. Its unusual vulnerability probably derives from its several unique features. The pulmonary capillary bed shows sequential arrangement, with a vast endothelial surface area (∼126 m 2 in adult humans). It the first capillary bed exposed to any postischemic blood. Not only is it oxygen rich (supportive of oxidative biology), but normally it is also uniquely highly enriched for blood leukocytes. Further, the leukocyte/endothelial interactions central to the inflammatory process in the lung may take place in these capillaries rather than in postcapillary venules as in other organs.
Ischemic Preconditioning
Short or pulsed minor episodes of I/R insult can initiate cellular survival adaptations that enable cells to forestall injury from a subsequent, more severe ischemic event. Long believed to comprise an adaptive response by individual involved cells, recent data suggest that remote effects may help direct preconditioning. Most animal and human studies of this have been directed at cardiovascular and cerebrovascular ischemia, for which mechanistic details vary. Generally, there is an early preconditioning effect evident only if the interval between the first (potentially preconditioning) brief ischemic event and the subsequent more severe (injurious) ischemic event is less than about 2 hours. If the interval is 24 hours, preconditioning effects are dependent on gene transcription and protein synthesis. Emerging data suggest that preconditioning effects in the brain may be derived from epigenetic changes.
The converse strategy of ischemic postconditioning has also been observed to help protect from I/R injury. Although much less well understood than preconditioning, this strategy would allow an after-the-fact, or at least a during-the-fact, maneuver to spare further tissue injury, an exciting possibility if it is sufficiently tractable to be used in clinical medicine.
I/R in human biomedicine
In human biomedicine, I/R is believed to explain the genesis of vascular and tissue injury in various conditions in which blood supply is interrupted. Examples include myocardial infarction, stroke, organ transplantation, angioplasty and thrombolytic therapy, cardiopulmonary bypass, ischemic acute renal failure, aortic cross-clamping, and vascular insults affecting liver and intestine. In addition, occurrence of nonseptic acute lung injury (ALI) and MODS can derive from underlying I/R occurring in the context of severe traumatic injury. In general, attribution to I/R in these disease states has resulted from recognition that the data attainable from humans are highly consistent with the extensive data collected from experimental animal models of specific vascular events. The perspective that clinical sickle disease also reflects an underlying I/R pathophysiology finds support in both experimental studies of sickle transgenic mice and clinical observations made in humans.
Sickle mice and experimental I/R
With the exception of sickle studies, experimental animal models typically have used abrupt interruption of the arterial supply to a given organ or segment thereof. This large vessel maneuver recapitulates only a few events in sickle disease (eg, pulmonary infarction, circle of Willis stroke). Yet the lessons derived from such nonsickle studies predict features that have been found to be characteristic of sickle mouse models even at ambient air, without artificially induced hypoxic stress. Multiple aspects of the baseline features of sickle transgenic mice are consistent with an underlying I/R genesis, as shown in Table 1 .
| Leukocytosis | Kaul et al, 2000 |
| ↑ Serum amyloid P | Belcher et al, 2003 |
| Oxidant stress | Osarogiagbon et al, 2000 Dasgupta et al, 2006 Aslan et al, 2007 |
| ↑ Lipid peroxidation | Dasgupta et al, 2006 |
| ↓ Plasma NO x levels | Dasgupta et al, 2006 |
| ↑ NFκB activation | Belcher et al, 2003 |
| ↑ Conversion of XD to XO | Osarogiagbon et al, 2000 |
| ↑ XO on endothelium | Ou J et al, 2003; Pritchard et al, 2004 |
| ↑ White blood cell/endothelial interaction | Kaul et al, 2000 |
| ↑ Platelet activation | Polanowska-Grabowska et al, 2010 |
| Endothelial cell activation | Solovey et al, 2001 Solovey et al, 2004 Aufradet et al, 2013 Vinchi et al, 2013 |
| ↑ HO-1 | Kaul et al, 2008 |
| Aberrant vasoregulation | Nath et al, 2000 Kaul et al, 2000 Aslan and Freeman, 2007 Kaul et al, 2008 |
I/R Induction in Sickle Mice
Sickle disease is understood to involve multifocal microvascular occlusions. Investigators have simulated this process by subjecting sickle mice to transient hypoxia, followed by reoxygenation by return to ambient air (hypoxia/reoxygenation [H/R]). This strategy is intended to statistically increase the chance of sickling and occlusion (with location[s] unknown), followed by some resolution thereof, within a defined time window that enables experimental observation.
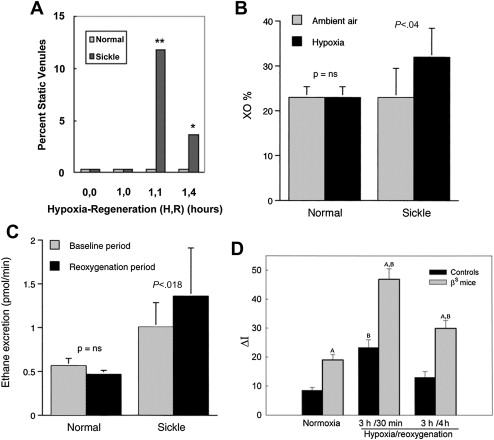
In the seminal sickle study, mild-phenotype sickle mice (NY1DD) were exposed to transient hypoxia (8% O 2 for 3 hours), which induced a 10-fold increase in number of sickled RBC. In this manner, several sickle models have been studied. On return to ambient air they showed (within the first 2 hours): vascular occlusions ( Fig. 1 A ); conversion of XD to XO (see Fig. 1 B ); and increased oxidant generation, including within endothelial cells (see Fig. 1 C , D ). In accordance with the expected I/R cascade, they developed increased NFκB activation in lung and liver ( Fig. 2 A ) and, after a delay, increased IL-6 and TNF-α in plasma (see Fig. 2 B ).
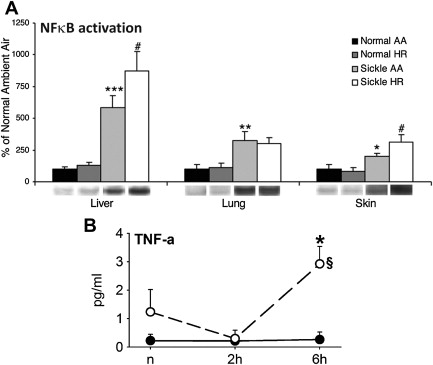
Some hours later, pulmonary endothelial cell activation became evident via upregulation of tissue factor and vascular cell adhesion molecule 1 (VCAM-1), with increased whole lung messenger RNA for VCAM-1, intercellular adhesion molecule 1 (ICAM-1), IL-1β, endothelin 1, eNOS, and tissue factor. In addition, there was a dramatic increase in (P-selectin–dependent) leukocyte/endothelial interactions, including transmigration ( Fig. 3 A–D ) and augmentation of pain behaviors, possibly mediated by mast cell activation.
Related posts:
 Does Erythropoietin Have a Role in the Treatment of β-Hemoglobinopathies?
The Role of Adenosine Signaling in Sickle Cell Therapeutics
Modulation of Hepcidin as Therapy for Primary and Secondary Iron Overload Disorders
Hemoglobin S Polymerization and Red Cell Membrane Changes
Ischemia-reperfusion Injury in Sickle Cell Anemia
Alterations of the Arginine Metabolome in Sickle Cell Disease
Does Erythropoietin Have a Role in the Treatment of β-Hemoglobinopathies?
The Role of Adenosine Signaling in Sickle Cell Therapeutics
Modulation of Hepcidin as Therapy for Primary and Secondary Iron Overload Disorders
Hemoglobin S Polymerization and Red Cell Membrane Changes
Ischemia-reperfusion Injury in Sickle Cell Anemia
Alterations of the Arginine Metabolome in Sickle Cell Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


