Targeted Agents in Squamous Cell Carcinoma of the Head and Neck
Jonas A. de Souza
Ezra E. W. Cohen
Everett E. Vokes
INTRODUCTION
Squamous cell carcinoma of the head and neck (SCCHN) is a heterogeneous disease of cancers located in different anatomic subsites and made up of diverse molecular biology. As our understanding of the molecular biology of the disease evolves, so do therapy options. This chapter will further describe the main mechanisms of disease, important pathways, and newer agents under investigation in SCCHN.
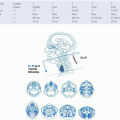
Over the last several years, epidemiologic studies have revealed that HPV-related oropharynx cancers represent a distinct disease entity based on clinical and molecular characteristics. HPV-related SCCHN is primarily driven by silencing of critical tumor suppressor genes by viral proteins and will be discussed in detail in Chapter 13. Several other oncogenes and tumor suppressors are dysregulated in non-HPV SCCHN and, for the first time, comprehensive exome sequencing data are available to catalogue these changes.1,2 Some important pathways are described in Figure 7-1. The full impact of these alterations on tumorigenesis, prognosis, and therapeutics has yet to be realized, but generalizations can be made regarding which pathways are most frequently disrupted in SCCHN.
Epidermal Growth Factor Receptor and Its Ligands
Epidermal growth factor receptor (EGFR) is a transmembrane receptor tyrosine kinase that is almost universally expressed in SCCHN. It contains an extracellular ligand-binding domain, a hydrophobic transmembrane segment, and an intracellular tyrosine kinase domain that is autophosphorylated upon activation. The primary ligands in SCCHN appear to be amphiregulin and transforming growth factor alpha (TGF-α), which induce homo- or hetero-dimerization with other receptors.3 EGFR activation has been linked with proliferation, invasion, angiogenesis, and metastasis. Moreover, increased EGFR expression has been associated with worse outcome in patients treated with surgery, radiotherapy, or chemotherapy.4,5,6
EGFR expression is primarily driven by gene amplification or polysomy, which together are observed in 40% to 50% of SCCHN.7 Increased EGFR expression appears even in preneoplastic lesions, and preclinical studies have demonstrated a reversal of SCCHN formation upon inhibition of EGFR.8 Therefore, EGFR has been a rationale target in SCCHN although protein expression, gene copy number, or ligand expression have not been reliable predictive parameters in patients treated with EGFR inhibitors.9
PI3K/AKT Pathway
The PI3K/AKT/mTOR pathway has been implicated in many cancers, and activation of this pathway contributes to formation and progression of SCCHN as well as resistance to chemotherapy and radiation. Constitutive AKT activation has been associated with amplification of the gene encoding the pllOa catalytic subunit of PI3K (PIK3CA) or AKT2; activating mutations in PIK3CA or AKT1; and PTEN loss. In studies of SCCHN tumor specimens, PIK3CA, AKT2, or PTEN alterations were found in 30% to 50%.10,11,12
p53
The p53 gene product is a pleiotropic protein that is the most commonly mutated gene in SCCHN (50%-60%).1,2 Functionally, p53 is involved in the regulation of diverse cellular processes including cell cycle, apoptosis, autophagy, DNA repair, and metabolism. Mutations generally render the gene nonfunctional and provide growth advantage to cancer cells. Specific p53 mutations have been linked with poor outcome in SCCHN and likely render tumors more resistant to therapy.13 Interestingly, p53 mutations are almost exclusively associated with HPV negative, tobaccorelated SCCHN.14
Cyclin D1
Entry into S phase of the cell cycle is dependent on phosphorylation of pRb by cyclin D1-cyclin-dependent kinase (CDK)-4, cyclin D1-CDK6 and cyclin E-CDK2 complexes. This leads to release of inhibition of key transcription factors that activate genes required for cell-cycle progression. Cyclin D1 itself is tightly regulated through transcriptional control (e.g., Myc), protein degradation (e.g., GSK3β), or inhibition of its partner CDKs by CDK inhibitors (e.g., p16). However, when cyclin D1 is amplified, which is detected in approximately 30% of SCCHN,15,16,17,18 physiologic controls can be overridden driving rapid proliferation and tumor growth. Cyclin D1 amplification has been associated with early carcinogenesis and resistance to therapy including cisplatin, EGFR targeting agents, and other small molecule kinase inhibitors.
With these important pathways in mind, an extensive list of agents has been developed and grouped in the following sections by their mechanism of action.
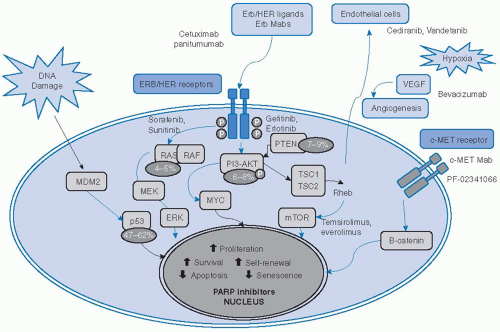 FIGURE 7-1. Selected pathways, targeted agents, and mutations important in squamous cell carcinoma of the head and neck. Red arrow, inhibitory activity; blue arrow, stimulatory activity; yellow circles; frequency of mutated genes; bold names, targeted drugs; PARP, poly(ADP-ribose)polymerases. (See color insert.) |
EPIDERMAL GROWTH FACTOR RECEPTOR INHIBITORS
Cetuximab, a recombinant chimeric anti-EGFR monoclonal antibody, was the first drug approved by the FDA for SCCHN since cisplatin, and it remains the only targeted therapy to have an indication and be routinely used in clinical practice for this disease. It inhibits activation of the receptor tyrosine kinase in addition to inducing antibody-dependent cellular cytotoxicity.19 Cetuximab is approved in combination with radiation therapy for locally advanced disease, in combination with platinum-based chemotherapy and 5-fluorouracil (5-FU) for the first-line treatment of recurrent/metastatic disease, and as a single agent for R/M disease after failure of platinum-based chemotherapy.
Vermorken et al.20 investigated cetuximab monotherapy in a phase II trial enrolling 103 patients with R/M SCCHN who failed to respond to platinum-based therapy. Patients were allowed to receive platinum plus cetuximab as salvage treatment upon disease progression. The best overall response rate—the primary endpoint of the trial—was 13% (95% confidence interval [CI], 7%-21%) with no complete responses seen. Of the 53 patients (51%) who received platinum plus cetuximab at progression, no responses were seen. Two phase II trials21,22 further tested the addition of cetuximab to cisplatin in patients who were refractory to platinum-based combination chemotherapy. In the first trial,21 the addition of cetuximab to platinum chemotherapy, the latter administered at the same dose and schedule at which progressive disease (PD1) was documented, produced an overall response rate of 10% in 96 patients, compatible to single agent cetuximab and likely only exposing patients to added cisplatin toxicities. The second trial22 enrolled 132 patients to receive two 3-week cycles with cisplatin/paclitaxel or cisplatin/5-FU. Patients (n = 30) with a complete or partial response continued standard therapy and cetuximab was given with cisplatin to those with stable disease (SD), PD1 and progressive disease within 90 days after platinum-based therapy (PD2). In total, 9 out of 51 patients who had SD after the first 2 cycles, 5 out of 25 patients who had progressed after 2 cycles of chemotherapy (PD1), and 3 of 54 patients who had progressed beyond 2 cycles up to 90 days (PD2) achieved an objective response, with median duration of response of 4.2, 4.1, and 7.4 months for the PD1, PD2, and SD groups, respectively, with median overall survival times of 6.1, 4.3, and 11.7 months. The lessons learned from these early trials were twofold: (a) cetuximab in combination with these agents was safe and tolerable at full doses of the respective cytotoxic agents; (b) the response rates of cetuximab as a single agent in platinum-refractory patients-suggest that its activity can be attributed mostly to the addition of cetuximab and that the monoclonal agent alone may achieve the same results. These trials led to a phase III trial in the first-line treatment for R/M SCCHN, conducted by the Eastern Cooperative Oncology Group (E5397).23 In a placebo-controlled randomized phase III trial of cisplatin (100 mg/m2 on day 1 every 4 weeks) with or without cetuximab (400 mg/m2) loading dose on week 1 followed by 250 mg/m2 weekly, a total of 117 patients with SCCHN were randomized. The patients had newly diagnosed metastatic disease or locoregional recurrence/persistence after initial locoregional therapy. A significant improvement in the objective response rate was
observed for the cetuximab plus cisplatin patient group, with 22.66% versus 9.3% (p = 0.03). Although the trial did not have the power to show significant differences in survival endpoints, the cisplatin-cetuximab arm had longer progression-free survival (4.2 vs. 2.7 months, hazard ratio [HR] for progression of 0.78; 95% CI, 0.54-1.12) and a trend for a longer median overall survival (9.2 vs. 8.0 months, p = 0.21).
observed for the cetuximab plus cisplatin patient group, with 22.66% versus 9.3% (p = 0.03). Although the trial did not have the power to show significant differences in survival endpoints, the cisplatin-cetuximab arm had longer progression-free survival (4.2 vs. 2.7 months, hazard ratio [HR] for progression of 0.78; 95% CI, 0.54-1.12) and a trend for a longer median overall survival (9.2 vs. 8.0 months, p = 0.21).
The definite trial was the Erbitux in First-Line Treatment of Recurrent or Metastatic Head and Neck Cancer (EXTREME), which confirmed the benefit of adding cetuximab to standard chemotherapy as a first-line treatment in the R/M setting.24 In total, 442 patients with R/M SCCHN, who were not candidates to local therapy and had not received systemic therapy in this disease setting, were randomized to treatment with either cetuximab plus platinum-based chemotherapy (cisplatin or carboplatin plus 5-FU) or platinum-based chemotherapy alone. Patients received cetuximab at a loading dose of 400 mg/m2 followed by 250 mg/m2 weekly until progression or unacceptable toxicity and either carboplatin (AUC, 5; Day 1) or cisplatin (100 mg/m2 intravenously; Day 1) plus 5-FU (1,000 mg/m2 intravenously; Days 1-4) every 3 weeks for a maximum of 6 cycles or the same dose and schedules of platinum plus 5-FU without cetuximab. Crossover of patients after disease progression was not allowed. The primary endpoint was OS with secondary endpoints of PFS, best overall response, disease control (complete response + partial response + stable disease), time to treatment failure, duration of response, and safety. The most common primary site in both arms was oropharynx (36% in cetuximab arm, 31% in chemotherapy alone arm).
The addition of cetuximab to platinum-based chemotherapy with fluorouracil (platinum-fluorouracil) significantly prolonged the median overall survival from 7.4 months in the chemotherapy-alone group to 10.1 months in the group that received chemotherapy plus cetuximab (HR for death, 0.80; 95% CI, 0.64-0.99; p = 0.04). The addition of cetuximab further prolonged the median progression-free survival time from 3.3 to 5.6 months (HR for progression, 0.54; 95% CI, 0.43-0.67; p < 0.001). The addition of cetuximab improved the response rate from 20% with chemotherapy alone to 36% with chemotherapy plus cetuximab (p < 0.001), although the duration of response did not differ significantly (4.7 months in chemotherapy arm, 5.6 months with cetuximab).
These clinical benefits were achieved with no changes in quality of life associated with the addition of cetuximab to chemotherapy.25 Of the 219 patients receiving cetuximab, 9% had grade 3 skin reactions and 3% had grade 3 or 4 infusion-related reactions. Patients in the cetuximab arm also had significantly more hypomagnesemia (p = 0.05) and sepsis (p = 0.02). Six patients in the cetuximab arm had grade 3 or 4 infusion reactions compared with none in the chemotherapy alone group. There were 10 treatment-related deaths, 3 on the cetuximab and 7 in the chemotherapy-alone arm, although there were no cetuximabrelated deaths. In a subset analysis, there was a greater benefit for patients below 65 years of age, those with better performance status, and those who received cisplatin (as three quarters of study participants did, and also compatible with data that suggest that carboplatin is not as effective as high-dose cisplatin.26 Based upon these data, cetuximab is approved for first-line use in combination with platinum-based chemotherapy in both Europe and the United States, and a triplet regimen consisting of platinum (preferably cisplatin), 5-FU and cetuximab should be strongly considered in symptomatic patients with a good performance status.
In the locally advanced setting, cetuximab is approved in combination with radiotherapy based on a multinational trial in which 424 patients with oropharynx, hypopharynx, or larynx locally advanced tumors were randomly assigned to radiotherapy with or without concurrent weekly cetuximab.27 Cetuximab was initiated 1 week before radiotherapy with a loading dose of 400 mg per square meter, followed by 250 mg per square meter weekly throughout radiotherapy.
Median overall survival of patients treated with cetuximab and radiotherapy was 49.0 (95% CI, 32.8-69.5) versus 29.3 (95% CI, 20.6-41.4) months in the radiotherapy-alone group (hazard ratio [HR] 0.73; 95% CI, 0.56-0.95; p = 0.018). Five-year overall survival was 45.6% in the cetuximab-plus-radiotherapy group and 36.4% in the radiotherapy-alone group. The addition of cetuximab to radiotherapy was associated with similar rates of grade 3 or 4 toxic effects in both groups. The incidence of acneiform rash and related hypersensitivity reaction was significantly higher in the combination cetuximab-radiotherapy group than in the group receiving radiotherapy alone. Rash and nail changes appeared to be the most common side effects associated with cetuximab. A further unplanned and underpowered subset analysis-showed that this benefit was limited to patients below the age of 65 and with a Karnofsky performance score (KPS) 90 to 100, and those with oropharyngeal cancer, findings that have to be confirmed.
Further, the interpretation of this trial is challenging since chemotherapy was not part of the study, making it difficult to assess whether the combination of cetuximab and radiotherapy was as effective as concurrent chemoradiotherapy. RTOG 0522 further enrolled 940 patients with locally advanced squamous cell carcinoma of the oropharynx, hypopharynx, or larynx to answer this question.28 Preliminary results from the clinical trial RTOG 0522 did not show benefits of concurrent cisplatin (100 mg/m2 on days 1 and 22) plus accelerated radiotherapy (70 Gy in 42 fractions over 6 weeks) with cetuximab when compared to concurrent treatment without cetuximab. With a median follow-up of 2.4 years, neither was there any improvement in progression-free survival, the primary endpoint of the trial, nor was there a statistically significant improvement in overall survival (83% vs. 80%, HR 0.87, 95% CI, 0.66-1.15). The final analysis of this trial is eagerly awaited.
Other monoclonal EGFR receptor inhibitors have also been investigated. Panitumumab is a fully humanized IgG2 monoclonal antibody with high affinity to the extracellular domain of EGFR,29 currently approved for use in metastatic colorectal cancer. Because of its structure (fully human antibody), infusion-related reactions are minimal. After promising early phase trials,30 a trial similar in design to the EXTREME trial was performed. The SPECTRUM trial31 assessed the addition of panitumumab to cisplatin and 5-FU chemotherapy had clinical activity with a significant increase in PFS and response rate, but failed to demonstrate a survival benefit. With median PFS of 5.8 months for patients in the panitumumab arm and 4.6 months for patients in the chemotherapy-alone arm (HR = 0.78; 95% CI, 0.66-0.92; p = 0.004), and overall response rate of 36% in the panitumumab arm, compared to 25% for patients who received only chemotherapy (descriptive p = 0.007), the median overall survival was 11.1 months in the panitumumab arm, compared to 9.0 months in the chemotherapy-alone arm (HR = 0.87; 95% CI, 0.73-1.05; p = 0.14). In a subsequently planned subanalysis, outcomes were analyzed by tumor HPV status. Of the 657 patients enrolled in the trial, 443 (67%) were evaluable for HPV testing by central review. There were 83 (22%) HPVpositive tumors and 294 (78%) HPV-negative tumors. In patients with HPV-negative disease, the addition of panitumumab was associated with significantly improved OS (11.7 vs. 8.6 months; HR = 0.73; 95% CI, 0.58-0.93; p = 0.01) and PFS (6.0 vs. 5.1 months; p = 0.001).32 However, this result has yet to be confirmed in further studies, given the lack of data that define HPV as a prognostic factor in R/M SCCHN or predictive factor regarding anti-EGFR therapy, as well as limitations in this analysis,
including its retrospective nature and missing HPV status in 35% of patients. Furthermore, similar findings were not seen in the phase II randomized CONCERT I trial of chemoradiotherapy with or without panitumumab in patients with unresected, locally advanced SCCHN.33 In this trial, compared with the cisplatin and radiation with regard to PFS and OS, there was a trend favoring concurrent cisplatin and radiation without panitumumab, with the addition of panitumumab increasing the HR of death to 1.63 (p = 0.1223).
including its retrospective nature and missing HPV status in 35% of patients. Furthermore, similar findings were not seen in the phase II randomized CONCERT I trial of chemoradiotherapy with or without panitumumab in patients with unresected, locally advanced SCCHN.33 In this trial, compared with the cisplatin and radiation with regard to PFS and OS, there was a trend favoring concurrent cisplatin and radiation without panitumumab, with the addition of panitumumab increasing the HR of death to 1.63 (p = 0.1223).
Zalutumumab, previously known as HuMax-EGFr, is a completely human IgG1 monoclonal antibody against human EGFR that also triggers Fc-mediated antibody-dependent cellular toxicity. Following a phase I/II study that included 28 patients with R/M SCCHN after failure to conventional treatments showed promising activity,34,35 an open-label phase III study was conducted. A total of 286 patients with SCCHN who were regarded as incurable by standard therapy and had PD1 within 6 months of platinum-based therapy were randomized in a 2:1 ratio to receive zalutumumab plus best supportive care (191 patients) or best supportive care with optional methotrexate (95 patients). Zalutumumab was dose-escalated to achieve a grade 2 rash. Patients in the best supportive care arm had the option to receive weekly methotrexate up to 50 mg/m2/week. Eligible patients were allowed up to two prior chemotherapy regimens in addition to treatment with platinum. There was a statistically nonsignificant increase in overall survival with zalutumumab compared with best supportive care (6.7 vs. 5.2 months; HR = 0.77; 95% CI, 0.57-1.05). The percentage of patients surviving at 12 months was longer with zalutumumab (22% vs. 12%). Zalutumumab significantly prolonged PFS from 8.4 weeks in the control group to 9.9 weeks (HR for progression or death, 0.63; 95% CI, 0.47-0.84; p = 0.0012). Response rate for zalutumumab was 6% including two complete responses. Grade 3 to 4 infection, hypomagnesemia, and cardiac events occurred more frequently in the zalutumumab group than in the control group. Given that methotrexate was utilized by 78% of patients in the control arm and that poststudy chemotherapy drugs other than methotrexate were given earlier and more frequently in the control group, the study essentially compared zalutumumab with an active treatment arm.
Small-molecule EGFR tyrosine kinase inhibitors have also been investigated in SCCHN. The most studied agents are erlotinib and gefitinib. Several trials assessed the single agent activity of gefitinib, an anilinoquinazoline reversible EGFR inhibitor that has been shown to be effective as a single agent in SCCHN cell lines and tumor xenografts as well as a radiosensitizer agent. Its promising early results have not translated into a survival benefit in a randomized trial. In the R/M setting, a randomized phase III trial of gefitinib versus standard methotrexate including 486 patients.36 Patients were stratified by whether they had received prior platinum-based chemotherapy (group A) or were too ill to receive such treatment (group B). Patients in each group were randomized to three treatment arms: oral gefitinib (250 or 500 mg daily) or intravenous methotrexate (40 mg/m2 weekly with dose escalation to 60 mg/m2 weekly) until disease progression. The overall response rates were 2.7%, 7.6%, and 3.9% in the gefitinib (250 and 500 mg) and methotrexate arms, respectively. The median overall survival was similar in the three treatment groups: 5.6, 6.0, and 6.7 months in the gefitinib (250 and 500 mg) and methotrexate arms, respectively. However, patients who received gefitinib experienced higher rates of tumor-related hemorrhage than those who received methotrexate, and gefitinib was associated with skin rash in 29% to 39% of patients and with diarrhea in 26% to 39%. When combined with conventional chemotherapy, also in the R/M setting, a phase III randomized trial of docetaxel plus placebo or docetaxel plus gefitinib 250 mg orally until disease progression was also reported.37 Although there was a statistically significant difference in time to progression from 2 months with docetaxel/placebo to 3.5 months with docetaxel/gefitinib (p = 0.03), there was no increased overall survival.
In the locally advanced setting, gefitinib was further incorporated into the chemoradiotherapy regimen in 67 patients.7 Two cycles of induction therapy with carboplatin and paclitaxel followed by FHX (5-FU, hydroxyurea, and radiotherapy on days 1 through 5 every 2 weeks for 5 cycles) and concomitant gefitinib 250 mg daily were administered, and gefitinib was continued for 2 years after the start of chemoradiotherapy. Grade 3 and 4 toxicities included mucositis (76% grade 3, 10% grade 4), dermatitis (29% grade 3, 3% grade 4), and diarrhea (1% grade 3, 0% grade 4). The majority of patients (n = 62) received maintenance gefitinib, and the median number of weeks on gefitinib was 95. The estimated overall 2-year survival was 83 %, with PFS of 77%. Disease-specific survival at 2 years was 86%. The overall clinical response rate was 91%. These data further suggested a potential role for EGFR inhibition in combination with standard chemoradiotherapy regimens. Furthermore, maintenance gefitinib was feasible and this finding may support future chemopreventive strategies with EGFR-inhibitors.
Erlotinib is an orally available, potent, reversible, and selective inhibitor of the EGFR tyrosine kinase. In a large phase II trial including 150 patients receiving erlotinib 150 mg daily,38 an overall response of 4.3 %, median OS of 6.0 months, and median PFS was 9.6 weeks were noted. Given these results, one can conclude that erlotinib as a single agent is marginally cytoreductive in this patient population. When combined with cisplatin in a phase I/II trial, erlotinib 100 mg daily and cisplatin 75 mg/m2 every 21 days39 showed a response rate of 21%, with one complete response, eight partial responses, and disease stabilization in 21 patients (49%; 95% CI, 33%-65%). Median progression-free survival was 3.3 months (95% CI, 2.7-4.8 months), and median overall survival was 7.9 months (95% CI, 5.6-9.5).
Another single arm phase II study assessed 60 previously untreated patients with stage III or IV locally advanced SCCHN (60% with oropharynx tumors).40 Patients received induction chemotherapy with 6 weeks of paclitaxel, carboplatin, infusional 5-FU, and bevacizumab; followed by concurrent chemoradiotherapy that included weekly paclitaxel, bevacizumab, and erlotinib. Radiation therapy began on day 1, with 1.8-Gy single daily doses, Monday through Friday, to a total dose of 68.4 Gy. Paclitaxel 50 mg/m2 was administered by 1-hour intravenous infusion on day 1 and weekly for six consecutive doses. Bevacizumab 15 mg/kg, intravenous infusion, was administered on days 1 and 22. Erlotinib 150 mg daily began concurrently with radiation therapy and continued daily during the 7-week course of radiation. At 2 and 3 years, the estimated progression-free survival rates were 83% and 71%, respectively, whereas the 2- and 3-year overall survival rates were 90% and 82 %, respectively. The investigators reported significant toxicities, with treatment breaks in 72% of patients, treatment noncompletion in 18%, two treatment-related deaths, and one myocardial infarction. In addition, 46 patients (76%) required enteral or parenteral nutrition during treatment. There are currently several clinical trials in progress assessing erlotinib for head and neck cancer. An interesting use of this agent has been in the chemoprevention setting. A clinical trial assessing erlotinib versus placebo as chemopreventive agents in a patient at high risk of developing SCCHN has finished accrual.
Lapatinib is an oral, small-molecule, reversible inhibitor of both EGFR and ErbB2. In a phase II trial in R/M SCCHN, oral lapatinib 1,500 mg daily was given to two groups of patients, those with and without previous exposure to an EGFR inhibitor.41 In all the evaluable patients, no objective responses were observed in either cohort, and lapatinib appeared to have no activity in this disease setting. In an intent-to-treat analysis, SD was observed in 37% of patients without prior exposure, and 20%
of those who had previously received an EGFR inhibitor. Thus, lapatinib appeared to have little clinical activity as a single agent in patients with SCCHN who were EGFR inhibitor naive or who had refractory disease. In the locally advanced setting, a phase II study including 107 therapy-naive patients were randomized (2:1) to receive 1,500 mg of oral lapatinib or placebo.42 During the short duration (14 days) of lapatinib treatment, there was a statistically significant reduction in mean tumor cell proliferation index in patients who had received lapatinib compared to the placebo arm (-6% vs. -1.7%, respectively, p = 0.030), and 17% had a complete or partial response, compared with no responders on the placebo arm. A further randomized phase II trial comparing lapatinib (1,500 mg/day) or placebo with concurrent chemoradiotherapy followed by maintenance therapy with lapatinib or placebo in unresected LAHNC43 showed a complete response rate at 6 months postchemoradiotherapy of 53 % with lapatinib versus 36% with placebo. Furthermore, a phase III study (NCT00424255) is in progress to assess the benefit of lapatinib (1,500 mg) or placebo once-daily with radiotherapy and cisplatin for 7 weeks in high-risk SCCHN patients.
of those who had previously received an EGFR inhibitor. Thus, lapatinib appeared to have little clinical activity as a single agent in patients with SCCHN who were EGFR inhibitor naive or who had refractory disease. In the locally advanced setting, a phase II study including 107 therapy-naive patients were randomized (2:1) to receive 1,500 mg of oral lapatinib or placebo.42 During the short duration (14 days) of lapatinib treatment, there was a statistically significant reduction in mean tumor cell proliferation index in patients who had received lapatinib compared to the placebo arm (-6% vs. -1.7%, respectively, p = 0.030), and 17% had a complete or partial response, compared with no responders on the placebo arm. A further randomized phase II trial comparing lapatinib (1,500 mg/day) or placebo with concurrent chemoradiotherapy followed by maintenance therapy with lapatinib or placebo in unresected LAHNC43 showed a complete response rate at 6 months postchemoradiotherapy of 53 % with lapatinib versus 36% with placebo. Furthermore, a phase III study (NCT00424255) is in progress to assess the benefit of lapatinib (1,500 mg) or placebo once-daily with radiotherapy and cisplatin for 7 weeks in high-risk SCCHN patients.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


