Introduction
T-cell development is a distinctive branch of hematopoiesis as well as a crucial input to immune system function. T-cell precursors, like other blood cells, are continuously generated from hematopoietic stem cells through a process that involves loss of access to alternative hematopoietic fates as well as gain of lineage-specific characteristics. In addition, like B cells, T cells must undergo programmed rearrangement of gene segments in order to assemble the genes that will encode their receptors for antigen, the α, β, γ, and δ T-cell receptor (TCR) chains. T-cell development is more distinctive in other ways. Unlike almost all other hematopoietic cell types, T-cell precursors must leave the bone marrow in order to adopt their fate. Most of them must migrate to the thymus for their differentiation: a specialized epithelial organ that provides a framework for intensive signaling that guides the cells to become T cells at the expense of other developmental fates. The T-cell program itself is also remarkable because of the variety of different types of T cells that it produces. Despite indelibly establishing common T-cell features, the cells that pass through T-cell development retain great functional plasticity and eventually fan out into a spectrum of different effector subtypes. Some of their diversification begins in the thymus, even while their universal T-cell properties are being acquired. Remarkably, as described in the following, some of the same transcription factors simultaneously play roles both in universal T-cell gene expression and in competitive oppositional mechanisms that determine particular effector subtype choices.
Thus, the T-cell program is more than a simple progression toward maturity: it is a mosaic of developmental processes that on the one hand forge a durable T-lineage commitment and on the other hand open up many different routes to useful T-cell functionality. T-cell function depends on recognition of antigen, and so a crucial part of the arming of the T-cell precursor for function is assembly of the TCR complex. To generate cohorts of T cells with various different recognition specificities, this process depends on individual cells’ random choice of TCR gene segments for rearrangement, and on additional chance incorporation of somatic mutations in the TCR genes, potentially different for every cell. The process is capable of generating autoreactive receptors or defective receptors as well as useful ones. Thus, the T-cell development program also incorporates several discrete filtering steps to eliminate newly made cells if they have dangerous or useless TCR specificities. These “repertoire selection” checkpoints are major landmarks in the T-cell development process, and they often provide choice points in developmental fate as well.
Both TCR recognition specificity and effector function subdivide T cells, and one of the most notable features of T-cell development is that particular functional subtypes become preferentially associated with particular specificities of antigen recognition. For one major divergence, the cluster of differentiation (CD) 4+ “helper” versus CD8+ “killer” decision, a gene network explanation has emerged, but explanations for differentiation of TCRγδ versus TCRαβ and “innate-type” versus “conventional” T cells of both receptor types remain a challenge.
This chapter covers T-cell differentiation both as a hematopoietic developmental program and as an immunological process that trains new cohorts of cells for appropriate discrimination between recognition of those targets that should elicit attack and those that should elicit protection. It begins with an introduction to the molecules that define T-cell identity and distinguish stages in the T-cell development process; a global synopsis of intrathymic T-cell development is then presented as a framework for the rest of the chapter. Subsequent sections zero in on particular steps in the process, developmental branch points, and checkpoints, in which crucial aspects of mechanism have been illuminated. The chapter then closes with a survey of some open challenges that are faced in the next few years.
Key Molecules for T-Cell Identity: Receptors, Stage Markers, and Effector Molecules
One goal of T-cell development is to arm mature T cells with a full set of the molecular criteria for T-cell identity. The mature T cell requires not only recognition apparatus, centered around the TCR complex, but also signal transduction apparatus to convey quantitative recognition information to the nucleus, and a poised transcriptional response system that can induce an appropriate combination of effector genes in response.
T-Cell Receptor Complex Components
In all jawed vertebrates, there are two forms of TCR complex: one with antigen recognition mediated by an αβ heterodimer and one using a γδ heterodimer. Individual T cells use either αβ or γδ receptors, but not both. All four chains, α, β, γ, and δ, are multidomain immunoglobulin superfamily proteins, type I transmembrane proteins (single-pass proteins
with N termini on the exterior of the cell). The highly variable N-terminal domain sequences of TCR chains mediate recognition of antigen, usually a peptide bound to a major histocompatibility complex (MHC) molecule on the surface of an antigen-presenting cell (APC). The variable domains are encoded by combinations of gene segments that assemble variably during a specific developmental phase when somatic mutation/recombination occurs. TCRα and TCRγ coding genes are assembled by joining segments of two types (V to J), while TCRβ and TCRδ genes are assembled by joining V, D, and J segments. Once the rearrangements have finished, the receptor sequences and recognition specificities of the T cells are fixed forever afterwards. As a rule, each T cell expresses only a single TCRα (or γ) and only a single TCRβ (or δ) chain. TCR heterodimer presentation at the cell surface then depends on assembly with a complex of signaling chains that do not themselves mediate antigen recognition, called CD3γ, δ, and ε and TCRζ (or CD3ζ). Like the TCR genes themselves, expression of the CD3 and ζ chains is T-cell specific and a result of gene activation induced by the T-cell developmental program.
T-Cell Receptor Coreceptors and Signal Modulators
TCR engagement with MHC-peptide ligands on APCs is greatly enhanced by coreceptors called CD4 and CD8 that interact with the most conserved parts of the MHC molecules. CD4, a linear, monomeric immunoglobulin superfamily molecule, interacts with class II MHC, while CD8, a dimeric molecule (either CD8αβ or CD8αα), interacts with class I MHC. The cytoplasmic domains of both molecules provide docking sites for a Src-family protein tyrosine kinase, Lck, that plays a key role in T-cell signaling. Thus, CD4 and CD8 not only stabilize TCR-MHC complex interactions but also ferry a major signaling mediator to the site of TCR-MHC interaction.
Lck activation upon TCR engagement triggers a complex cascade of signaling pathways. While some of the pathway components are universal mediators of calcium, PI3-Kinase, Ras/MAP kinase, and NF-κB activation, T-cell responses also use specialized components such as the adaptor LAT, protein kinase Cθ and the T-cell-specific kinases Zap70 and Itk. In addition, other cell surface receptors also feed into these pathways to modulate the signals. One signal-amplifying receptor is CD28, an immunoglobulin superfamily disulfide-linked dimer. Another transmembrane signaling molecule, CD5, is induced by TCR signaling and appears to provide feedback signal-damping functions. These molecules are not strictly T-cell specific but are tightly developmentally regulated within the T-cell pathway.
Activation Markers/Developmental Stage Markers
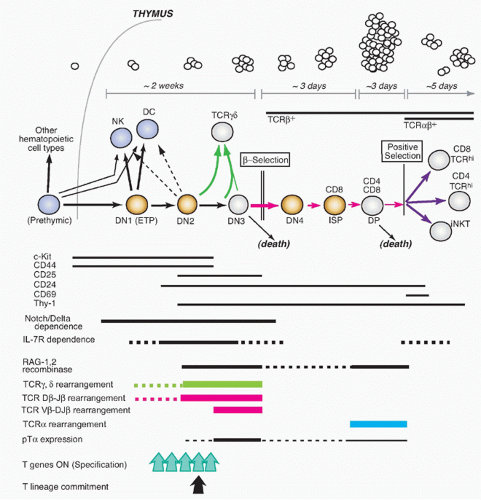
When T cells are activated, three additional cell-surface molecules are usually upregulated with different kinetics. CD69 is usually first, expressed transiently, followed by CD25 and then by CD44, which is the most persistently expressed. CD25 can be used as a subunit of the receptor for the growth factor IL-2 (as IL2Rα) but can also be expressed alone. In mature T cells, persistent CD44 expression marks memory and antigen-experienced effector cells, whereas CD25 is most often a marker for Foxp3+ Treg cells. However, all three markers have different roles in T-cell development, where their expression patterns usefully distinguish various stages.
Thy-1, a glycolipid-linked membrane receptor, is highly expressed in all murine T-lineage cells from an early stage on, though not in human. CD24 or “heat-stable antigen,” another cell surface molecule expressed in many hematopoietic precursors, is highly expressed in all TCR-negative stages, gradually downregulated in a stepwise fashion throughout T-cell development, and finally turned off when intrathymic maturation is complete.
Growth Factor Receptors
Despite the importance of interleukin (IL)-2 and IL-4 in mature T cells, complete IL-2 and IL-4 receptors are not required for most aspects of T-cell development. However, receptors for IL-7 are crucial for the initial expansion of T-cell precursors in early stages of T-cell development, indispensable for TCRγδ cell development, and probably also important for divergence of CD4+ (helper) and CD8+ (killer) branches of TCRαβ cell development. At the earliest stages of T-cell development, IL-7R function is anticipated and supplemented by Kit, which is the receptor for “stem cell factor” (steel factor, Kit ligand), that is also essential for early T-cell development. Kit and IL-7R are dynamically regulated with distinct patterns of expression across early stages of T-cell development.
Effector Response Molecules
Mature T cells are armed for particular stereotyped responses to antigen recognition. Most CD4 helper cells make their first antigen response by turning on expression of the growth factor IL-2. Killer T cells respond to activation by expressing the membrane-perforating molecule perforin, granzymes A and B, and relatively toxic cytokines like interferon (IFN)γ. Th1 cells do so by expressing IFNγ and TNFα, but much less perforin. Th2 cells respond by expressing IL-4 and in some cases IL-5 and/or IL-13. Natural killer T (NKT) cells rapidly activate IL-4 expression together with IFNγ. Th17 cells, often found defending epithelia, express inflammatory cytokines such as IL-17A, IL-17F, and IL-22. Additional subsets, follicular helper T cells and Th9 cells, also have particular canonical effector programs that they use to respond to TCR signals. Whereas all these subtypes promote immune activation upon stimulation, regulatory T cells (Treg cells) respond to stimulation by blocking the effector responses of neighboring T cells, at least in part through expression of IL-10, TGFβ, and specific cell-surface interaction molecules.
In mature T cells, the poised state of distinct sets of cytokine genes that distinguishes each subset is maintained, in between rounds of stimulation, by transcription factor expression patterns and site-specific chromatin modification. Particular transcription factors provide distinctive, crucial regulatory inputs in particular cell types. Thus, Runx3, T-bet, and eomesodermin are important to maintain killer function, T-bet to maintain Th1 function, GATA-3 to promote Th2 function, PLZF to promote NKT-cell function,
RORγt to promote Th17 function, Bcl6 for follicular helper T-cell function, and Foxp3 to promote T
reg function. Of particular interest is the fact that most of these factors also play pivotal earlier roles in the generation of T cells in general.
The differentiation of particular effector subsets is usually considered to start with antigen-inexperienced (naïve) mature T cells, after they have finished with their intrathymic development. The specialization itself is part of the mature T cell’s portfolio of possible responses to antigen, especially among CD4 T cells. Conventionally, one may consider these antigen-driven events to be a completely different type of developmental process from the intrathymic events that mold hematopoietic precursors into T cells. However, we will see in the following that effector programming can also originate in the thymus for subsets like NKT cells, certain classes of TCRγδ cells, and some Tregs: the distinction is not absolute.
Narrative Summary: Major Stages of Intrathymic T-Cell Development
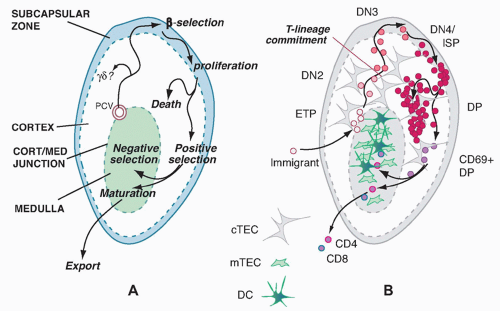
The major known site for T-cell development in all jawed vertebrates is the thymus.
1 The thymus does not harbor its own long-term source of precursors but is continuously populated throughout life by new precursors that are replaced as their descendants complete maturation and selection. These precursors migrate to the thymus from bone marrow or fetal liver when they are still multipotent. The thymus not only enforces their commitment to a T-cell fate and T-lineage differentiation but also provides a proliferation-inducing environment. It is estimated that the numbers entering the thymus per day are quite modest, on the order of < 100 per day for a typical young adult mouse.
2,3 The descendants of that small cohort of input cells expand over a 2-week period to a cohort of cells that have undergone T-lineage commitment, TCR gene rearrangement, and TCR expression, eventually yielding ˜50 × 10
6 new thymocytes a day before their proliferation stops.
Major Subdivisions Based on CD4, CD8, and T-Cell Receptor-αβ Status
The steps of T-cell differentiation in the mouse thymus are summarized in
Figure 13.1.
3,4,5,6 Important landmarks for the process are provided by the expression patterns of the TCR coreceptors, CD4 and CD8, and the timing of rearrangement of the TCR-coding genes themselves.
Cells in the early stages of development in the thymus are CD4- CD8- (“double negative” [DN]) and cannot yet express TCR. While the DN stages can be subdivided (described subsequently), the cells in all the DN stages are mostly determining their commitment to a T-cell fate, proliferating, and preparing for their first expression of TCR complexes. With successful expression of a TCRβ chain, they turn on CD4, CD8α, and CD8β genes together. The resulting CD4+ CD8αβ+ “double positive” (DP) cells are highly distinctive to the thymus, not found among mature T cells in peripheral lymphoid organs, and are diagnostic of a pivotal stage in T-cell development and survival. The DP cells in steady state constitute by far the major fraction of thymocytes (˜80%), and many of them come to express complete TCRαβ receptors as well. However, the overwhelming majority of these cells are fated never to go further in their differentiation. Only a small fraction are allowed to progress further through positive selection, to become either CD4+CD8- TCRαβ+ cells (“CD4 cells”) or CD4- CD8αβ+TCRαβ+ cells (“CD8 cells”). It is estimated that the cells have only about 3 days within the DP stage in which to achieve this success before their window of opportunity closes. Left in the cortex, the other DP cells of their cohort die “of neglect,” to be replaced by progeny of the next cohort of precursors, at turnover rates up to 50 × 106 per day.
T-Cell Receptor-αβ Rearrangement and Selection Checkpoints
The regulated process of TCR rearrangement and selection helps to explain why so many DP cells must be produced. TCR coding genes are assembled through a highly regulated but highly imprecise recombination process mediated by the recombinase complex RAG1/RAG2, the same recombinase that rearranges immunoglobulin VDJ gene segments in B cells. The imprecision is useful to add to the diversity of the eventual T-cell recognition pool, but it also results in many gene rearrangements that are out of reading frame or otherwise defective. To winnow out the useful cells, TCR gene rearrangement occurs in two sequential bursts, each followed by a quality control checkpoint at which all the cells with defective receptors can be eliminated.
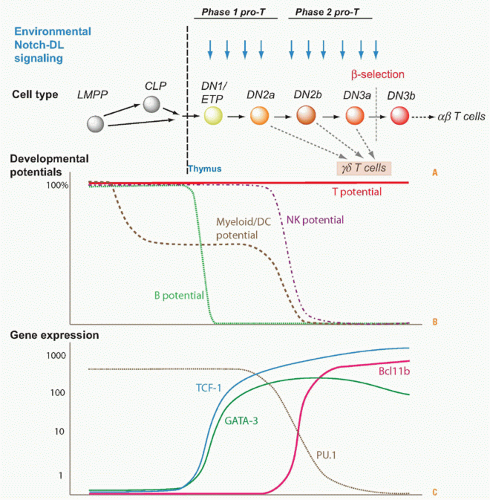
The timing of different phases of recombination competence is determined by the specific activation and deactivation of the RAG1 and RAG2 gene products; the genetic loci on which their effects can be focused at any given time are determined by developmentally regulated unmasking via localized chromatin opening. For TCRαβ cells, the most numerous T cells in mice and humans, the TCRβ gene must rearrange first, while the cells are still in the DN stage. The cells carrying out this rearrangement need to pause proliferation in order to allow the RAG complex to work, and most often they are never allowed to divide again unless they succeed in generating a good TCRβ coding sequence through rearrangement at one allele. This strict condition is the “β-selection checkpoint.” Only cells passing this checkpoint are allowed to become DP cells, receiving a bonus of multiple rounds of cell division to expand the winners and dilute the stalled, dying losers. Through the process of β-selection, then, the cells also shift the gene loci that are accessible for rearrangement, losing the ability to rearrange their TCRβ genes (and TCRγ and TCRδ genes) any further and acquiring the ability to rearrange their TCRα genes.
Once they become DP cells, TCRα rearrangement gets underway. As a successful in-frame TCRα chain rearrangement occurs, the heterodimer TCR complexes of the new TCRα chains with the previously generated TCRβ chains become the next subject of testing at a second checkpoint. This second checkpoint is more draconian than the first, as the criterion for success here is not simply successful expression of a TCRαβ heterodimeric protein, but also the quantitative details of the recognition specificity of the new
receptor, as measured by its interaction with ligands in the thymic environment. The newly expressed TCR could fail to recognize anything, making it useless, or it could confer on the cell a dangerous autoreactivity that could lead to autoimmune disease. All of these failures must be eliminated before they appear in the mature T-cell population. The only successful cells are the ones that happen to have heterodimeric receptors with the right combination of functionality and low affinity for self-ligands. These are the ones that are allowed to become CD4 or CD8 cells through a process termed “positive selection,” with the CD4 cells generally becoming cytokine-producing “helpers” and the CD8 cells generally becoming “killers.” The positively selected cells permanently silence their RAG gene expression and undergo further maturation steps but little if any additional cell division before they are sent out to the periphery.
The effects of the ordered TCR gene rearrangements and quality control checkpoints are seen clearly in the effects of different mutations on the progression of T-cell development.
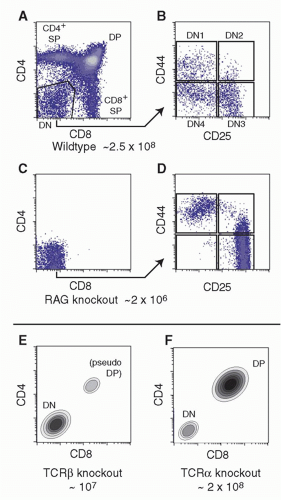
Figure 13.2A,B depicts the CD4/CD8 profiles and DN subsets in a normal young mouse thymus of about 2 to 3 × 10
8 cells.
Figure 13.2C,D shows the effects on thymus populations in mutant mice in which TCR gene rearrangement is impossible (eg, via mutation of
Rag1 or
Rag2 or
Prkdc, the gene that encodes a deoxyribonucleic acid [DNA]-dependent protein kinase needed for rejoining of the gene segments during rearrangement [a very instructive mutant,
Prkdcscid, causes severe combined immune deficiency]). Because TCRαβ-lineage cells are so predominant in the mouse thymus, the effect of mutations of
Tcrb alone appears very similar
(Fig. 13.2E). In contrast, mutations of the
Tcra locus alone allow production of a full complement of DP cells, although CD4 and CD8 SP cells are completely missing
(Fig. 13.2F).
T-Cell Receptor-based Lineage Diversification for T-Cell Receptor-αβ Cells
The first functional distinctions between TCRαβ cell subsets apparently arise only during emergence from the DP stage. Positive selection itself probably triggers these divergences through distinctive modes of positive selection signaling, as we discuss in detail in a later section. The three main outputs of TCRαβ cell positive selection are CD4 cells, CD8 cells, and NKT cells, so called because they share some functional qualities and surface receptors with natural killer (NK) cells. DP cells first experiencing positive selection signals immediately turn on the activation marker CD69, start increasing the surface density of their newly validated TCRαβ complexes, upregulate CD5, and then begin the process that results in shutting off either CD8αβ or CD4 to generate TCRαβ-high “CD4 single positive” and “CD8 single positive” cells, respectively. As the cells arrive at one of these “single positive” (SP) phenotypes, they still retain expression of another marker of immaturity, the CD24 “heat-stable antigen” that is expressed from the early DN stages onward. However, they turn off both CD69 and CD24 as they reach completion of their functional maturity, in a process that takes 3 to 7 more days.
7 NKT cells undergo a distinctive maturation sequence that is discussed separately in the following, but they too lose CD69 and CD24 as they mature.
The selection of these different subsets is based on TCR and coreceptor interactions with different ligands in the thymic environment. CD4 cells are positively selected by TCR and CD4 joint interaction with MHC class II molecules. CD8 cells are positively selected by TCR and CD8αβ joint interaction with MHC class I molecules. NKT cells are positively selected by nonclassical class I MHC molecules, using non-CD4, non-CD8 accessory molecules (signaling lymphocytic activation molecule [SLAM] family molecules) to assist in signal triggering. Although the primary trigger in each case is TCRαβ-ligand recognition, the effector programs of these different major lineages diverge significantly, through the consequences of using these different types of coreceptors to assist in “interpreting” the TCR signals. How this signal-dependent feature of T-cell maturation works is a major focus of later sections of this chapter.
Negative Selection of Autoreactive T-Cell Receptor-αβ Cells
Many cells with TCR that might be dangerously autoreactive can be eliminated at the DP stage, but another crucial period for this aspect of quality control is after positive
selection. As newly generated single-positive cells display increased surface density of their TCRαβ, raising their avidity for MHC-bound ligands, those that have strongly autoreactive receptors are generally killed off by interaction with specific subsets of thymic stromal cells in a process called “negative selection.” This continual purging of the newly made T-cell recognition repertoire is crucial to avoid autoimmune disease, as important mouse mutations and human disease models reveal. An alternative pathway to defeat autoreactivity, at least among CD4+ cells, is to alter the functional subtype of the autoreactive cells, to program them for function as tolerogenic regulatory T cells before they even have a chance to leave the thymus. Generation of such “natural T
regs” (nT
regs) is but one case where TCR interaction properties direct the cells to a particular T-cell functional class.
Generation of T-Cell Precursors in the Double Negative Compartment
For years, the least well understood set of thymocytes was the DN cells. The DN-stage cells comprise only a small fraction of thymocytes in steady state, generally less than 5%, but in fact represent the generative compartment of the thymus in which the largest number of total cell cycles takes place. Profound developmental transformations also occur during transit through the DN stages.
Understanding the basis of T-cell specification became possible only through the discovery of markers that could subdivide the DN compartment into successive stages (reviewed in refs. 2,3). The earliest stages of precursor differentiation within the thymus are represented by DN (CD4- CD8- TCR- or sometimes CD4low) cells that express high levels of the growth factor receptor Kit and the activation/adhesion molecule CD44 but low levels of CD25, termed Kit-high DN1 cells or early T-cell precursors (ETPs). Then, these cells turn on expression of CD25, signaling their definitive entry into the T-cell pathway, and the resulting Kit+ CD44+ CD25+ cells are called “DN2” cells. Both the DN2 and ETP stage cells proliferate, and IL7R is increasingly expressed in the DN2 stage. The DN2 cells then progress into the “DN3” stage (CD25high but now CD44low and Kitlow), and here proliferation slows or stalls, RAG protein levels rise, and efficient TCRβ rearrangement can proceed. Development terminates at the DN3 stage unless the cells can pass β-selection or γδ-selection.
Successful TCRβ expression and passage of β-selection enable the cells to restart proliferation, shut off expression of CD25, and pass through a transitional “DN4” or “preDP” stage (CD25low, CD44low, Kitlow). They then proceed to acquire CD8 and CD4 on the cell surface, sometimes CD8 slightly before CD4 (“immature SP”), and finally culminate their differentiation into DP cells by finishing their proliferation (large DP to small DP transition). This extended β-selection-dependent transition from DN3 to small DP cells creates a unique regulatory state in the resulting DP cells that prepares them for selection. Notably, although some of these cells may be positively selected as described previously, only a minority may ever divide again until after they have left the thymus.
Distinctive Paths for T-Cell Receptor-αβ Cells
T cells that will use TCRγδ instead of TCRαβ actually follow a special program or set of program options from the DN2 stage on. Whereas TCRβ rearranges strictly before TCRα and mostly in DN3 stage, the TCRγ and TCRδ genes can rearrange in parallel within the DN2 or DN3 stage. Thus, a RAG1/RAG2+ cell can in some cases acquire successful inframe rearrangements of both TCRγ and TCRδ genes before it is successful with TCRβ alone. As a result, newly TCRγδ+ cells arising within the DN2 or DN3 population leave the TCRαβ lineage mainstream, undergoing a γδ-selection process that is different from β-selection. The γδ-selected cells generally keep CD4 and CD8 silent, shut off RAG1/RAG2 permanently without promoting TCRα rearrangement, and limit their proliferation to a much smaller number of cell cycles than β-selected cells.
It is not clear yet whether all TCRγδ cells must go through two sequential checkpoints analogous to β-selection and positive selection. Like TCRαβ cells, TCRγδ cells are now understood to emerge in multiple functional subtypes, and we will discuss in a separate section possible ways that these subtypes become differentially programmed.