5.1 Introduction
An entirely new form of immunity, adaptive immunity, first evolved around the time of the Cambrian explosion. The first true lymphocytes appeared when fish developed jaws around 450 million years ago and adaptive immunity exists in all jawed vertebrates. Adaptive immunity has evolved in the presence of innate immunity, and depends in large part on the utilization of innate components for its activation and effector functions. In mice and men, innate immunity has now become largely subservient to the adaptive form in defence against infection. Why an additional form of immunity evolved is unknown, although presumably it could reflect an “arms race” between infectious agents and their hosts. Nevertheless, the ability of lymphocytes to recognize infectious agents may have been facilitated by mobile DNA elements (“jumping genes”) which are present in complex organisms. Stable insertion of such elements may have contributed to the evolution of a recognition system based on rearranging gene segments to generate multiple different antigen receptors. This system is used by T and B lymphocytes in vertebrates, and ultimately enables “anticipatory” recognition of antigens expressed by infectious agents.
In this chapter, we examine immune responses that are primarily dependent on T lymphocytes. We start by noting that different populations of T cells have evolved, before focusing mainly on one conventional type, the αβ T cell (Section 5.1). We introduce classical major histocompatibility complex (MHC) molecules, which co-evolved with T lymphocytes and that enable them to recognize cells containing infectious agents, and describe how these molecules work (Section 5.2). (We also note that some related types of non-classical MHC molecules can enable non-conventional T cells to recognize and respond to infection.) We next examine the responses of conventional T cells, where they are activated and how they are triggered (Section 5.3), and what is known of how they can subsequently develop specialized functions that are needed to eliminate different types of infectious agents (Section 5.4). After this we discuss the role of the thymus in T cell development, including the generation of a huge repertoire of antigen receptors by random DNA changes – which is essential for the anticipatory recognition of infectious agents (Section 5.5). We also note how non-conventional T cells also develop within in the same organ. Finally we turn to therapy and briefly describe how T cells can be used in vaccines against infectious disease and potentially against malignant tumours (cancers) (Section 5.6).
5.1.1 T Cell Populations
The primary division of T cells into different populations is in terms of the molecular nature of their antigen receptors. T cell receptors (TCRs) are composed of two chains, both of which contribute to the antigen recognition site. The best understood and most studied are the conventional αβ T cells, where α and β refers to the two chains of their antigen receptors. Later we outline some features of γδ T cells and invariant NKT (iNKT) cells (not to be confused with natural killer (NK) cells; Section 1.4.5). It is important to appreciate that the development of all these different populations of lymphoid cells occurs in the thymus, which is relatively shielded from the rest of the body, and that they are generated before any infection occurs. Their subsequent effector responses are, however, generally dependent on the prior sensing of infectious agents, particularly by components of innate immunity. See Figure 5.1.
Figure 5.1 T cell populations. The same T cell precursor can give rise to several T cell subsets with different functions. Conventional αβ T cells develop into CD4 and CD8 populations in the absence of foreign antigens. When they become activated by antigen they can develop further into different effector subsets (e.g. Th1, Th2 and perhaps Tc1, Tc2 cells, etc.) or into memory T cells. Some can also develop into regulatory T cells (not shown). Non-conventional T cell populations include γδ T cells and iNKT cells. (There are also non-conventional αβ cells that express neither CD4 nor CD8, and different types of NKT cells; not shown.)
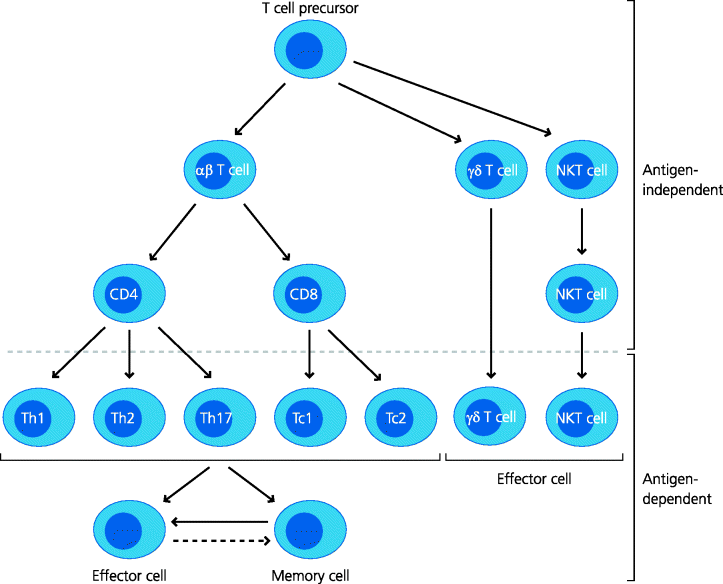
5.1.2 How Do Conventional T Cells Recognize Antigens?
The αβ T lymphocytes have evolved primarily to interact with cells containing antigens derived from infectious agents. In contrast, B cells can directly recognize antigens that are soluble or form part of an infectious agent. Many pathogens that have infected mammals spend much of their life inside host cells; these include all viruses, many bacteria and some protozoa. To function effectively, the adaptive immune system needs to detect and deal with such infections. This poses two problems for T cells – how can they recognize that they are interacting with another cell, and how can they detect that that cell is infected? The adaptive immune system has evolved a sophisticated mechanism to limit T cell activation to situations where the T cell is in contact with another cell. This is the primary function of the classical MHC molecules. These MHC molecules act as receptors for small peptides that are generated within different cellular compartments. In turn, the TCRs of conventional T cells recognize parts of bound peptides and the MHC molecules to which they are bound. This means that a single T cell shows dual antigenic specificity; both for the peptide bound to the MHC molecule and for the MHC molecule itself. The specificity of a T cell for a particular MHC molecule is known as its MHC restriction. We now know that this is because each chain of any given αβ TCR has three particular regions that differ most from those of any other TCRs (i.e. they are hypervariable) and which determine complementarity between the receptor and its ligand; hence they are termed complementarity-determining regions (CDR1–3 for each chain, respectively). For each, CDR1 and 2 primarily interact with exposed parts of the MHC molecule while CDR3 makes contact with the bound peptide. (Antibodies also exhibit similar hypervariable regions, but in these cases they only contact the antigen itself; Chapter 6.) See Figure 5.2.
Figure 5.2 Interactions of T cell receptors with peptide–MHC complexes. αβ TCRs recognize peptides bound to MHC molecules. These peptides are generated from proteins within cells by mechanisms collectively known as antigen processing and bind to MHC molecules before they are exported to the plasma membrane. The TCR α and β chains each contain three hypervariable CDRs (CDR1–3). The CDR3 regions interact primarily with the peptide while the others interact predominantly with the MHC molecule itself.
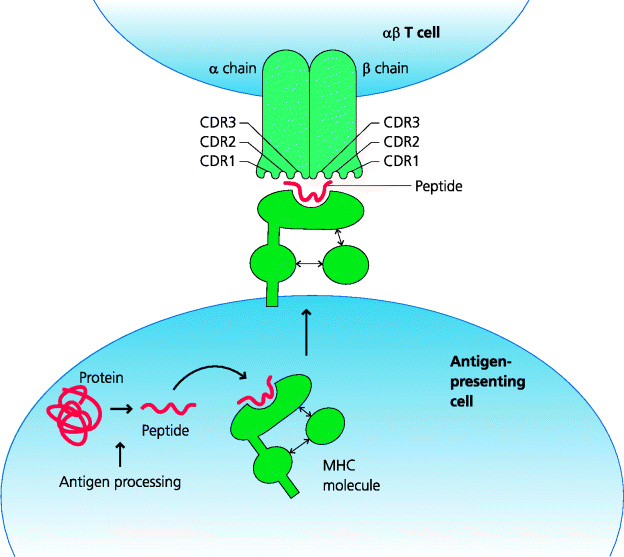
5.1.3 Subpopulations of Conventional T Cells
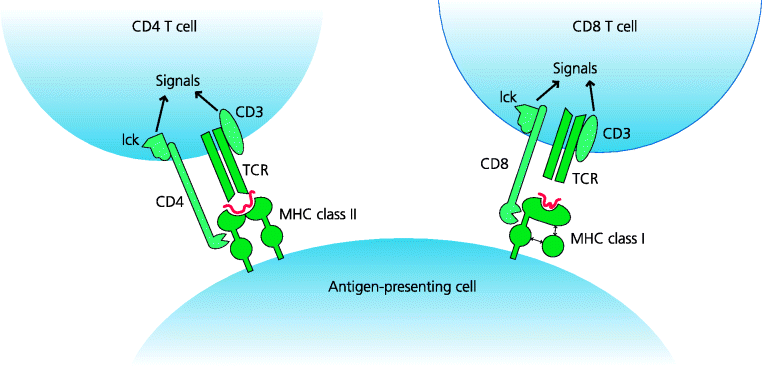
Conventional αβ T cells are divided into two main subpopulations: CD4 and CD8 T cells. The CD4 molecule binds to MHC class II molecules, whereas CD8 binds to MHC class I. This binding generates signals that contribute to T cell activation and is crucial in determining the different functions of the subsets. MHC class I is expressed on almost all nucleated cells. Expression of MHC class II is, however, restricted to a limited number of cells involved in CD4 T cell development, activation or effector functions. These include thymic epithelial cells, dendritic cells (DCs), B cells and some macrophages. See Figure 5.3.
Figure 5.3 Interactions of CD4 and CD8 with MHC molecules. CD4 and CD8 molecules are first expressed on thymocytes during T cell development, and which one of these a T cell eventually expresses determines whether it will interact with MHC class I or II. CD4 and CD8 bind to invariant parts of MHC class II and class I respectively. The TCR is associated with CD3, a multimolecular complex (not shown). CD4 and CD8 are constitutively associated with the tyrosine kinase, Lck, which phosphorylates specific regions ITAM motifs of CD3 (not shown) in the early stages of T cell activation.
5.1.4 Effector Functions of T Cell Populations
In this chapter we focus particularly on conventional CD4 and CD8 T cells. In general, CD4 T cells are the cells that regulate adaptive immune responses. For example, they can instruct B cells to develop into plasma cells that make different types of antibodies, recruit different types of granulocytes to specific sites of infection and regulate how macrophages function. They are also primarily responsible for many clinically important diseases or conditions, including allergies and autoimmune diseases, much forms of transplant rejection, and much of our inability to reject tumours (Chapter 7). CD8 T cells are generally thought to have less of a role in immune regulation, but are certainly crucial effector cells in both immunity to infection and in a number of important immune-mediated diseases.
5.1.5 T Cell-Mediated Immunity Against Infection
What is the evidence that we need T cells in defence against infectious disease? There is strong clinical evidence for this. For example, some children are born without thymuses (DiGeorge syndrome). These children and others with T cell deficiencies suffer from an increased incidence of viral infections. If such an individual is accidentally infected with vaccinia virus in smallpox vaccination, rather than the normal short-lived self-healing lesion, the infection spreads and may destroy large areas of the skin. Similarly, children who were genetically T cell-deficient (i.e. they suffered from a primary immunodeficiency disease) and who were immunized with Bacillus Calmette-Guérin (BCG) during vaccination programmes became infected and developed a widespread, potentially fatal infection. In acquired T cell deficiencies, such as acquired immunodeficiency syndrome (AIDS), a similar increase in susceptibility to tuberculosis is also seen. In addition, AIDS patients may become infected with strains of mycobacteria such as Mycobacterium avium, or with Pneumocystis jirovecii and cytomegalovirus (CMV), which are harmless to normal individuals. This can only mean that in normal individuals, the adaptive immune system, particularly the T cell system, is continually acting to recognize and eliminate such microbes without there being any clinical signs. There is also abundant experimental evidence for the crucial importance of T cells in defence against infection. See Box 5.1
5.2 Major Histocompatibility Complex (MHC) and Antigen Presentation
5.2.1 Classical MHC Molecules
It is not possible to understand how T cells work without understanding the role of the MHC. The function of conventional MHC molecules is to act as peptide-binding receptors that are recognized by antigen receptors on T cells. The MHC was discovered from early transplantation studies. It was known long ago that transplants between different members of the same species (allografts) were destroyed (rejected) very rapidly (Section 1.6.5). Early experiments led to the identification of genetic regions that controlled the rejection of such transplants. These were termed histocompatibility loci. One locus in particular controlled the most rapid rejection of these transplants and was thus termed the major histocompatibility locus. This locus was subsequently found to include a large complex of genes and became known as the MHC. In mice, the MHC is known as H-2 (because historically it was the second locus to be identified from transplant studies). In humans, it is known as HLA (short for human leukocyte antigens which were originally identified using panels of antibodies).
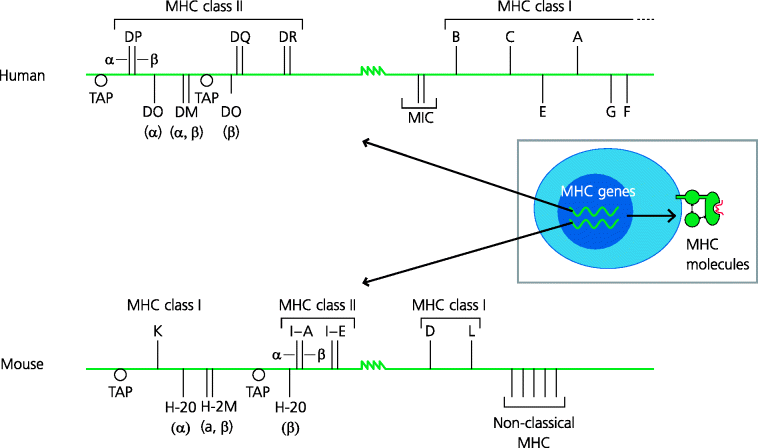
The MHC contains two different sets of genes that code for the classical MHC peptide-binding molecules. These are the MHC class I and class II loci and both contain several discrete structural genes. In humans, the MHC class I region contains three structural genes: HLA-A, -B and -C. In mice, the equivalents are H-2K and H-2D. The human MHC class II genes are called HLA-DP, -DQ and -DR, and in mouse they are called I-A and I-E. See Figure 5.4.
Figure 5.4 Genetic organization of human and mouse MHC. The MHC consists of a large stretch of DNA, in humans called HLA and in mice H-2. Several distinct structural genes encode the α chain of classical MHC class I molecules (which associate with β2-microglobulin, encoded outside of the MHC), and the α and β chains of class II molecules. Many other molecules are also encoded in the MHC region. Some of these are involved in peptide delivery to, or loading of, classical MHC class I or class II molecules; these, respectively, include TAP, and in humans HLA-DM and -DO (which are also dimers; A, B); the mouse equivalents are H-2M and H-2O. There are also non-classical MHC molecules, such as MIC(-A and -B), and HLA-E, with specialized functions.
MHC moleucles are codominantly. Thus, any individual is likely to express six different MHC class I molecules, each binding a different set of peptides. Another most important feature of the classical MHC loci is their extreme polymorphism. Thus, within human and mouse populations there are often more than 100 variants (alleles) of some structural MHC genes. Many other genes have multiple allelic variants in populations, but usually these variants are rare. The MHC is different in that most alleles are present with significant frequencies. The importance of this polymorphism is that it makes it difficult for a pathogen to escape recognition by a T cell both in an infected individual and in a population. The result is that if a pathogen mutates so a peptide from it can no longer bind to any given MHC molecule, there are five other MHC molecules that could potentially bind other peptides from the pathogen. At a population level, the vast numbers of MHC alleles available means that even if any pathogen mutates so that no peptides can be recognized by T cells of a particular individual, it is highly unlikely that any other individual will express the same set of alleles and thus most individuals within the population will remain resistant. The importance of this polymorphism is illustrated by African cheetahs. Cheetahs in captive colonies are susceptible to widespread infections. These animals suffered a reproductive bottle-neck at some time in their evolution, resulting in them having a very limited MHC diversity and this may explain their susceptibility. See Box 5.2
5.2.1.1 How Do Peptides Bind to Classical MHC Molecules?
The amino acid sequence of MHC molecules was determined by biochemical techniques but this did not reveal how they interacted with peptides. Many theories existed until X-ray crystallography showed the three-dimensional structure of a MHC class I molecule. This revealed that the outer part of the MHC molecule contained a groove into which a single peptide could be bound. A similar structure was identified for MHC class II molecules.
MHC molecules consist of two chains. In MHC class I molecules the heavy chain is a trans-membrane protein consisting of three domains and it wholly contains the peptide-binding groove. The heavy chain is associated non-covalently with a smaller molecule, β2-microglobulin, which represents the fourth domain, but this is encoded outside of the MHC. MHC class II molecules are formed from two chains, α and β, both of which are trans-membrane molecules and both of which contribute to the peptide-binding site. Structurally, the domains of MHC molecules are members of the immunoglobulin superfamily.
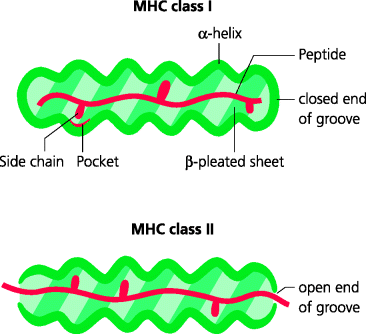
It was long known that peptides added to cells in culture could enable recognition by specific T cells, but the nature of the peptides that were naturally bound to MHC molecules was unknown. To resolve this, MHC molecules were isolated from cells and, by altering the ionic conditions, peptides were eluted from the MHC molecules and their characteristics determined by chemical or physical approaches (e.g. sequencing or mass spectrometry). These experiments illustrated several important features of MHC peptide interactions. Thus, peptides bound to MHC class I molecules tend to be 8–10 amino acids in length (typically 9), whereas those bound to MHC class II molecules can be considerably longer. The reason for this is that in MHC class I molecules the peptide-binding groove is closed at both ends, constraining the length of peptide that can be accommodated. In MHC class II molecules the groove is more open, permitting peptides of greater lengths to bind. See Figure 5.5.
Figure 5.5 Interactions of peptides with MHC molecules. The peptide-binding grooves of MHC molecules consist of two stretches of α-helix which overlie a β-pleated sheet base. Most of the differences (polymorphisms) between MHC molecules are found in the groove and hence determine precisely which peptides can be bound by different MHC alleles. Class I molecules have grooves closed at both ends, restricting the size of peptides that can bind. They also contain pockets into which side chains from the peptide amino acids can bind non-covalently as anchor residues to hold the peptide in place. Class II molecules have more open ends, allowing peptides of greater and more varied lengths to bind.
Peptides eluted from a particular MHC molecule are very heterogeneous in overall composition. However, it was found that, for MHC class I molecules, amino acids of similar types were present at particular positions in the eluted peptide (2 or more positions in each peptide) suggesting that these amino acids might be involved in anchoring the peptides to the MHC molecule. In fact, pockets in the peptide groove were subsequently identified from X-ray crystallographic studies. Binding of peptides to MHC class II also involves anchor residues although these tend to be more dispersed along the groove. Hence specific residues of the respective MHC grooves are crucial for high-affinity binding of peptides. Comparison of different MHC alleles also confirmed that most of the polymorphic residues are situated in the walls and base of the groove, thus determining which peptides can bind to any given MHC molecule. What is equally important is that because only relatively small number of residues are involved in binding to MHC class I molecules, for example, the remaining residues can be highly variable. Hence, a single MHC molecule can bind a multitude of different peptides, probably numbering many tens of thousands, making them highly promiscuous receptors.
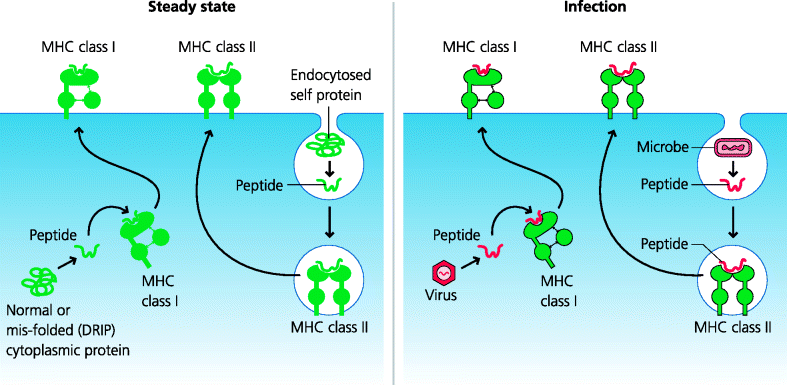
Clearly, MHC molecules are capable of binding peptides from infectious agents. But what do they bind in the absence of infection? When MHC-bound peptides were identified and sequenced, many of them were found to derive from normal cell-associated proteins. MHC class I-associated peptides are mainly derived from intracellular proteins (nuclear and cytoplasmic), whereas MHC class II peptides come from external proteins (such as those present in the culture media of cells) and also from proteins from the plasma membrane and endocytic compartments of the cell itself. What this suggests, and indeed what we now know to be the case, is that peptides from infectious agents in the cytoplasm (such as viruses) are likely to bind primarily to MHC class I molecules, while those from endosomal compartments (such as phagocytosed bacteria) will mostly bind to MHC class II molecules. See Figure 5.6.
Figure 5.6 Origins of peptides bound to MHC molecules. Under steady-state conditions, peptides bound to MHC class I molecules are mostly derived from abnormally-folded cytosolic proteins (defective ribosomal products; DRIPs). In contrast, peptides bound to class II molecules may originate from extracellular proteins that have been endocytosed or from plasma or endosome membranes. During infection peptides from viruses or microbes that have gained access to the cytoplasm, or which have been endocytosed, may become loaded onto either MHC class I or class II molecules, respectively.
5.2.1.2 Cellular Distribution of Classical MHC Molecules
In humans, apart from some central nervous system (CNS) neurons, all nucleated cells express MHC class I; erythrocytes, which lack nuclei, do not. MHC class II molecules show a much more restricted distribution. Under steady-state (resting) conditions, the cells in rodents that express MHC class II are DCs, B lymphocytes, thymic epithelial cells and epithelial cells in the small intestine. In humans a wider range of cells, including macrophages and activated T cells, may be MHC class II-positive. However, during active immune responses interferon (IFN)-γ can stimulate expression of MHC class II on many different cell types and can additionally increase the expression levels of MHC class I molecules.
5.2.2 Non-Classical MHC Molecules
Other molecules are encoded within the MHC region that are structurally similar to those of the classical MHC, but which differ functionally. These are non-classical MHC molecules. They mediate a diverse range of functions. For example, HLA-E (Qa-1 is similar in mice) is a non-polymorphic molecule. It binds leader sequence peptides from newly synthesized MHC class I molecules; these sequences target the MHC molecule to the rough endoplasmic reticulum (RER)). Signal sequences bound to HLA-E can be recognized by NK cells and are important in inhibiting NK cell killing. Two other non-classical MHC molecules include human MIC-A and MIC-B (which do not appear to bind peptides) that become expressed by injured or stressed cells, and may then be recognized by non-conventional γδ T cells (Section 5.4.5) as well as NK cells. Other molecules are also structurally similar to MHC molecules, but are encoded outside of the MHC region. Perhaps the best known are CD1 molecules which are able to bind lipid-containing molecules such as phospholipids and glycolipids, and which may be involved in anti-bacterial defence because they can be recognized by non-conventional T cells such as iNKT cells (Section 5.2.8.2).
5.2.3 The Cellular Basis of Antigen Processing and Presentation
It was known from very early studies that antibodies could bind directly to protein antigens in their natural three-dimensional confirmation but not when the protein had been denatured (i.e. if its tertiary three-dimensional structure was destroyed). However, when the activation of T cells was first studied it was clear that their antigenic requirements were very different. If T cells were removed from an immunized animal and stimulated in vitro with the antigen alone they did not respond. If, however, as well as antigen, other cell types such as macrophages were added to the cultures, the T cells became activated. It was soon found that the macrophages could be pre-cultured with the antigen and still activate the T cells in the absence of free antigen. It was also shown that macrophages could present denatured proteins, but needed to be metabolically active to present native or denatured proteins. Metabolically inactivated macrophages could, however, still activate T cells if peptide fragments from the antigen were added. The concept thus arose that antigens needed to be “presented” to T cells by other cells, rather than the T cells being able to recognize them directly. The antigen also needed to be processed, before it could be recognized. These are the origins of the terms antigen processing, antigen presentation and antigen-presenting cells (APCs). Now of course, as we introduce above and further discuss below, we have a clear mechanistic explanation for these early findings, but the terms have stuck.
What are APCs? Historically this term was first used for any cell that could activate helper (CD4) T cells. In contrast, studies of killing by CD8 cytotoxic T cells generally used cells called target cells. Hence a distinction came about between the concept of an APC (as a cell that expressed MHC class II molecules for CD4 T cell recognition) and a target cell (as a cell that expressed MHC class I molecules for CD8 T cell recognition). The later discovery of the crucial roles of DCs in activating naïve CD4 T cells led to further muddying of the waters. Cells that could activate naïve T cells were called professional APCs. Unfortunately there is still a general tendency to limit the use of these terms to CD4 T cell activation, although we now know that DCs are also often essential for CD8 T cell activation.
Even though we now have a clearer understanding of the molecular basis of peptide generation and loading onto MHC molecules, and of T cell activation by APCs, it is still convenient to use the terms antigen processing and presentation. Perhaps, however, the term APC should be used for any cell that expresses MHC molecules and which can be recognized by any conventional T cell. Thus, a cell expressing MHC class I can become an APC for a CD8 T cell and a cell expressing MHC class II can become an APC for a CD4 T cell. As mentioned, amongst APCs it is important to distinguish those APCs that can activate naïve T cells. These professional APCs are primarily DCs. Sometimes B cells and macrophages are included in this term, but there is little evidence that these cells can activate (or at least initially trigger) naïve T cells in vivo.
5.2.4 Antigen Processing and MHC Class I Presentation
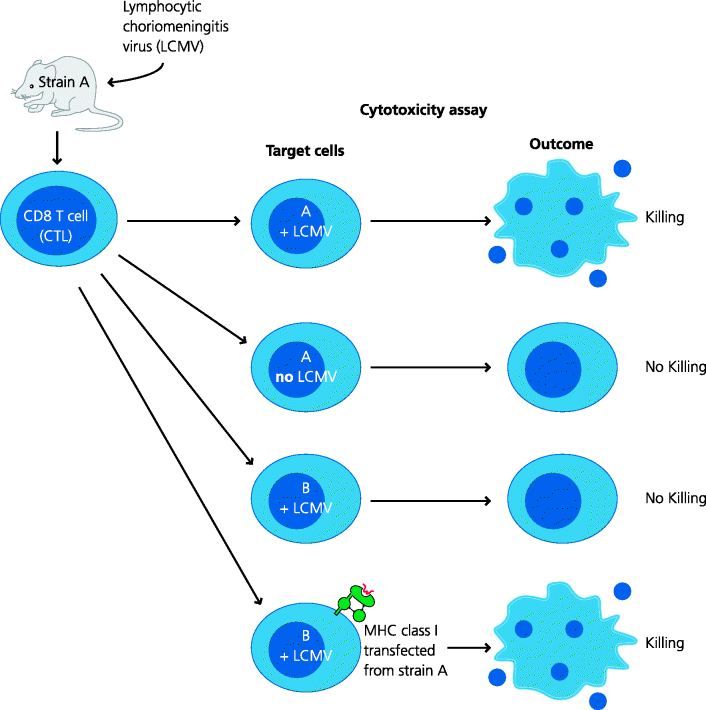
CD8 T cells were first characterized as cells that can kill other cells after they have been activated, and we now know that MHC class I molecules are crucial for recognition. How was this discovered? Doherty and Zinkernagel found that mice infected with lymphocytic choriomeningitis virus (LCMV) developed CD8 T cells that could kill virally infected mouse target cells in culture. However, for killing, it was essential that the target cells shared at least one MHC class I molecule with the infected mouse. This was the origin of the concept of MHC restriction which led to the award of a Nobel Prize to Doherty and Zinkernagel. Subsequent experiments by many groups showed that CD8 T cells recognized short peptides derived from cytoplasmic proteins and that these peptides were bound to MHC class I molecules. Generally speaking, peptides derived from the cytoplasm, whether or not they originate from self proteins or infectious agents (e.g. viruses or microbes that have infected cells), are termed endogenous antigens. In general, endogenous peptides are presented to CD8 T cells by classical MHC class I molecules. See Figure 5.7 and Box 5.3.
Figure 5.7 MHC restriction of CD8 cytotoxic T cells. Mice infected with many viruses generate CD8+ CTLs. These can be isolated and are able to kill cells in culture that are infected with the same virus. Doherty and Zinkernagel, using a particular virus, LCMV, found that only cells of the original mouse strain could be killed (e.g. A), while cells from other strains (e.g. B) could not even if they were infected with the same virus. Eventually they discovered that, if infected cells were to be killed, they needed to express the same MHC class I molecules as the mouse that was initially infected. This became known as MHC restriction. This principle applies to both CD8 and CD4 T cells: the former are restricted to MHC class I molecules, the latter to MHC class II.
5.2.4.1 The Proteasome
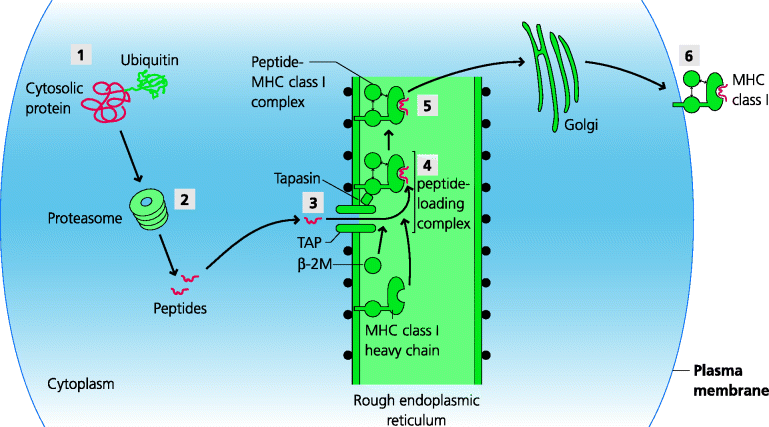
Endogenous peptides bind to MHC class molecules, but how are the peptides generated? Here we have an example of the adaptive immune system using a pre-existing mechanism from another system. Proteasomes are large, multi-protein complexes which degrade proteins into peptides and are part of normal cellular housekeeping, degrading proteins into short peptides for subsequent recycling of their constituent amino acids. Many proteins are targeted to proteasomes after they have been modified with multiple copies of another small protein, ubiquitin. Proteasomes consist of a stack of ring-like protein complexes, rather like a stack of American doughnuts. These form two smaller chambers at each end with a larger, central chamber with multiple proteolytic activities. Proteins are unfolded (by mechanisms we do not fully understand) and passed into the antechamber, then into the central chamber where they are cleaved, and peptides emerge at the other end. In response to IFN-γ the structure of the housekeeping proteasome is changed to form the immunoproteasome, which has different proteolytic activities. These tend to generate peptides that are better targeted to the MHC as they have C-termini that are good anchors for MHC class I molecules in one pocket of the groove. How do we know that proteasomes are important in processing for MHC class I? Lactacystin is a bacterial product that is a selective inhibitor of proteasomal activity and cells treated with lactacystin show inhibition of processing of some, but not all proteins. Hence, other cytoplasmic proteolytic mechanisms must exist. An important example is the ER-resident aminopeptidase-1 (ERAP1) enzyme that can cleave that N-terminal region of peptides, sometimes generating ends that fit well into an other pocket of the groove. See Figure 5.9.
Figure 5.8 Cellular localization of proteins recognized by CD8 cytotoxic T cells. Different strains of influenza virus express different forms of a relatively small number of proteins. One of these is the nucleoprotein (NuP) that is never expressed on the surface of infected cells. Mice were infected with one strain of the virus and their CTLs were tested for the ability to kill infected target cells in culture. The target cells were infected with strains of virus that shared different variant proteins with the original virus used to infect the mice. In some cases it was found that in order to render the target cells sensitive to killing, the only protein that needed to be identical in the two viruses was NuP. At the time this was a real puzzle. However, we now understand that the NuP protein is degraded intracellularly and peptides from it are about to MHC class I molecules and transported to the cell surface where these complexes can be recognized by CTL.
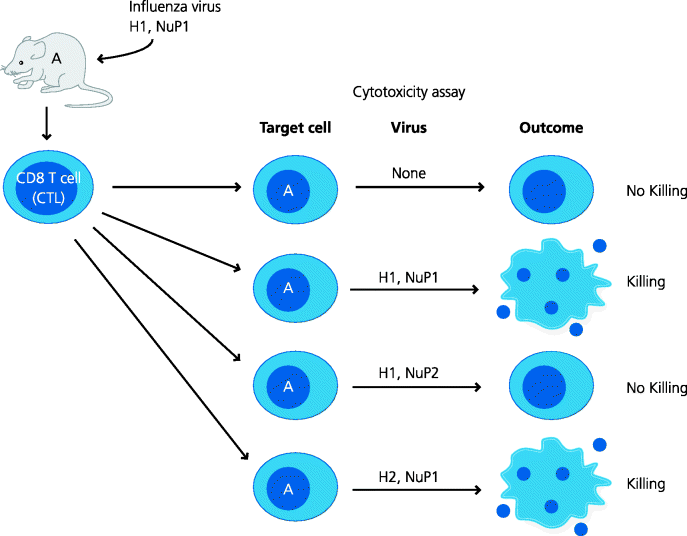
Figure 5.9 The peptide–major histocompatibility complex class I pathway. (1) Cytosolic proteins, often attached to ubiquitin, (2) are targeted to proteasomes (modified as immunoproteasome; no shown) where they degraded. (3) Peptides of appropriate sizes are transported into the RER by ATP-dependent TAP transporters. (4) These are associated, via tapasin, to newly assembled MHC class I molecules, with associated β2-microglobulin, to form the peptide-loading complex. Suitable peptides can bind to the MHC molecules, (5) which are then released and (6) transported through the Golgi apparatus to the plasma membrane. (The peptide-loading complex also contains chaperone proteins; not shown.)
5.2.4.2 The TAP Transporter
MHC class I-binding peptides are generated in the cytoplasm, but the MHC molecules themselves are synthesized in the RER, separated from the cytoplasm by a membrane. How can the peptides get across this membrane? They are transported by a bi-molecular complex, TAP, which is expressed in the RER and transport is ATP-dependent. TAP is selective in the sizes of peptides it can transport and for peptides that have particular types of terminal amino acids the fit better into MHC class I grooves. The TAP transporter belongs to a family of molecules specialized to transport a variety of small molecules across membranes (ABC transporters), so yet again the adaptive system has adapted a pre-existing system for its own ends.
5.2.4.3 The Peptide-Loading Complex and Peptide Editing
Once the peptide has been transported into the RER it may bind to a MHC class I molecule. Newly formed MHC class I molecules are attached to TAP by a complex including a molecule called tapasin to form the peptide-loading complex. Other molecules associated with this complex act as chaperones to control the retention, stability and structure of the MHC class I molecules. Tapasin may also increase the probability of the MHC molecule meeting transported peptides because the spectrum of peptide–MHC complexes expressed in tapasin gene knock-out mice and wild-type mice is different. Further modification of the peptide may also occur via enzymes present in the RER such as ERAP1. If in the end a stable peptide–MHC complex is formed, it is transported out of the RER through the Golgi apparatus to the cell surface. It is important to appreciate that peptide editing occurs throughout the MHC class I peptide-loading pathway, from immunoproteasomes, through TAP and tapasin, to help bias the spectrum of peptides towards those that are best able to bind to MHC class I molecules, sometimes by mechanisms which are still unclear. Peptide editing also occurs at different stages of the MHC class II pathway (below).
5.2.5 Antigen Processing and MHC Class II Presentation
Around the time that Zinkernagel and Doherty were investigating immune responses to viruses (above), other groups were examining the genetic basis of the immune response to proteins in mice. To simplify the experiments they used synthetic polymers of small numbers of amino acids. When different strains of mice were immunized with these artificial antigens they could be classified as high or low responders in terms of the antibody responses they made on secondary stimulation. Again, classical breeding experiments showed that a small number of genes controlling responsiveness were coded for in the MHC. These were originally called immune response (IR) genes and their products therefore became known as IR-associated (Ia) antigens. They are now known to be classical MHC class II antigens. We might explain these early findings at the level of the APC, on the basis of whether or not the synthetic polypeptides were able to bind to the particular alleles of MHC class II molecules. However, we could also explain these results at the level of the T cell. Since thymic MHC class II molecules control the repertoires of T cells produced in different strains of mice (Section 5.5.2), they could determine whether or not the T cells that develop can recognize these peptide–MHC complexes.
5.2.5.1 Endosomal Degradation of Foreign Antigens
Peptides that bind to MHC class II molecules are generated in the endosomal pathway, in contrast to those that bind to MHC class I which originate in the cytosol. Most cells have mechanisms for taking up and degrading extracellular proteins in the endosomal pathway, and the adaptive immune system uses these mechanisms for the generation of MHC class II-binding peptides. Proteins are taken up by fluid-phase or receptor-mediated endocytosis, or by phagocytosis, and degraded by lysosomal proteases in the progressively-acidified vesicles of the endosomal pathway. The importance of lysosomal proteases was shown in experiments in which protease inhibitors were added to cultures of APCs with whole proteins and it was found that MHC class II presentation was inhibited. Lysosomal proteases act at low pH, and if the endosomal pH was raised by adding weak bases such as chloroquine, again presentation was inhibited. DCs and perhaps B cells do show some specialization in terms of the proteases used and their regulation, but the principles are very much the same whatever the cell type involved. Generally speaking, peptides that originate from the endosomal pathway, whether or not they derive from self proteins or infectious agents such as phagocytosed bacteria, are known as exogenous antigens. Generally, exogenous antigens bind to classical MHC class II molecules and are presented to CD4 T cells.
5.2.5.2 Peptide Loading in the MHC Class II Pathway
Exogenous proteins are broken down in the endocytic pathway, but how do they become bound to MHC class II molecules? MHC class II molecules are synthesized in the RER; however, unlike many proteins, MHC class II molecules do not travel directly to the plasma membrane. They need to interact with peptides and these peptides are present in the endosomal pathway. MHC class II molecules are therefore targeted to the endocytic pathway and this is a function of the invariant chain with which they are initially associated. The invariant chain is synthesized in the RER and forms a complex with newly synthesized MHC class II molecules. A targeting sequence in the cytoplasmic tail of the invariant chain directs the complex via the Golgi apparatus into the endosomal pathway, where it can sample peptides in different parts of the pathway. See Figure 5.10.
Figure 5.10 The peptide–major histocompatibility complex class II processing. Proteins in the endocytic pathway are broken down to peptides by lysosomal proteases. Meanwhile, (1) MHC class II α, β and invariant chains are synthesized and (2) assembled in the RER. (3) The invariant chain both blocks the peptide-binding groove in the MHC molecule and targets the complex, through the Golgi, to the endocytic pathway. (4) Here, the invariant chain is digested in a step-wise manner to leave a small fragment, CLIP, which blocks the groove. (5) CLIP then either dissociates spontaneously or dissociation is facilitated by a MHC class II-related molecule, human HLA-DM (or mouse H-2M), which additionally promotes peptide exchange until a high-affinity peptide binds into the groove. (6) The peptide–MHC complex can then be transported to the plasma membrane.
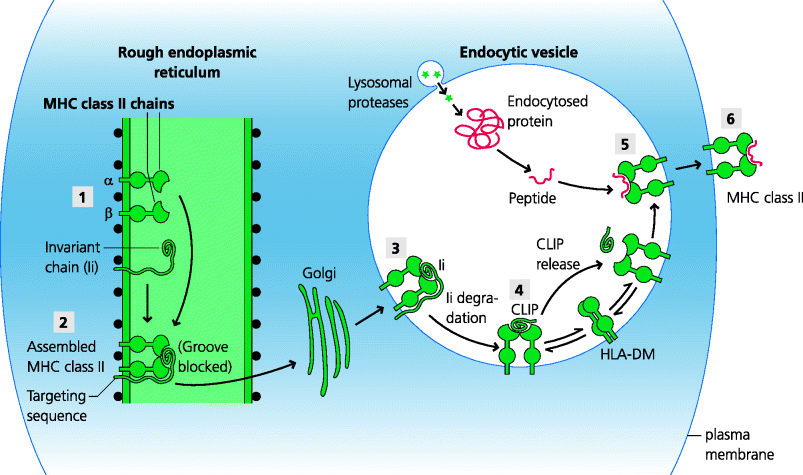
Another crucial function of the invariant chain is to block the peptide-binding grooves of MHC class II molecules. This is important because it ensures that peptides delivered into the ER for binding to MHC class I molecules (above) are unable to bind to newly synthesized class II molecules, and hence ensures these pathways are functionally discrete. However, once the class II molecules are delivered to the maturing endosomes, proteases start to cleave the invariant chain at specific sites until a single portion (CLIP) remains and continues to block the peptide-binding groove. Clearly peptides from foreign antigens cannot bind to class II molecules until this is released. Sometimes this can happen spontaneously. In other cases, however, CLIP release requires a different, MHC class II-related molecule – HLA-DM in humans or H-2M in mice – that is restricted to the endosomal pathway. In fact, this class II-related molecule (which is only present in endosomes) probably plays a more general catalytic role in peptide selection for binding to the groove, perhaps by maintaining a more open conformation of the peptide-binding groove until a high-affinity peptide binds. The peptide-bearing MHC class II molecule can now be transported to the plasma membrane where it can potentially be recognized by CD4 T cells. In some cells, particularly B cells and some DCs, the function of HLA-DM or H-2M is regulated by another class II-related molecule, HLA-DO in humans or H-2O in mice. See Box 5.4
5.2.6 Cross-Presentation
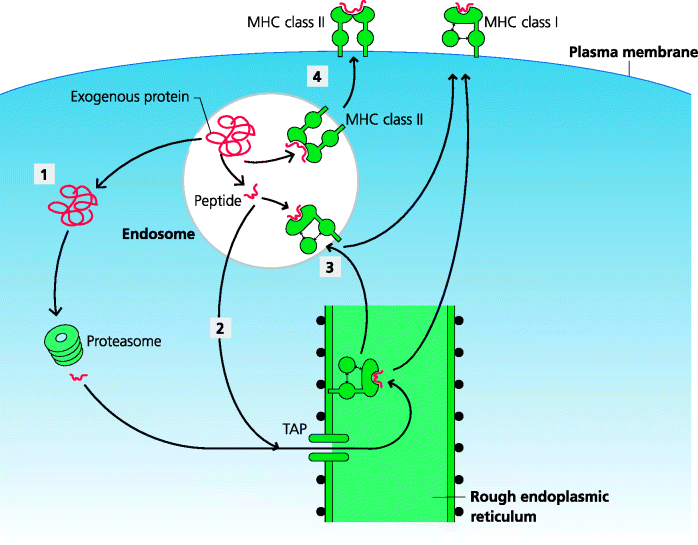
Most viruses are very selective in terms of the cell type(s) they can infect (tropism, Section 2.2.2.1). We know from depletion and adoptive transfer experiments that CD8 T cells are crucial for recovery from many viral infections, and also that CD8 T cells recognize MHC class I-peptide complexes derived from cytoplasmic proteins. We also know that, generally speaking, DCs are the principle, if not the only APCs that activate naïve T cells effectively in vivo (Section 5.4.1). This raises a real problem. How can a DC present antigens from all the different viruses that might infect a given species? It might be that DCs can be infected by all viruses; however, given viral tropism, this would imply that DCs need to express all the surface molecules used by viruses as receptors which is clearly very unlikely. At least one subset of DCs has, however, evolved a different solution to this problem. These cells can deliver exogenous peptides derived from the endosomal pathway (which would normally be delivered to class II molecules) onto MHC class I molecules instead. This process is known as cross presentation. This means that DCs can activate CD8 T cells to viruses that do not actively infect the DCs. Although several mechanisms have been proposed to explain cross-presentation, this is still an area of controversy. It has, for example, been suggested that, for this to occur, phagocytosed antigens need to be delivered into specialized endosomes and that soluble antigens, internalized by receptor-mediated endocytosis, might be targeted into one pathway or the other depending on the actual receptor involved. In some cases whole proteins, and in others peptides appear to be transported from the endocytic pathway into the cytosol. See Figure 5.11.
Figure 5.11 Cross-presentation. In some DCs, peptides from endocytosed proteins can be displayed on MHC class I molecules. This is known as cross-presentation. The mechanisms are still not fully understood. (1) Proteins or (2) peptides may exit the endocytic pathway into the cytosol. (3) Alternatively, endosomes may acquire MHC class I, possibly from the RER, permitting direct peptide binding. (4) Cross-presentation enables peptides derived from microbes in peripheral tissues to be presented on MHC class I molecules to CD8 T cells, without the need for the DCs to be actively infected. Such DCs can also present exogenous peptides on MHC class II, additionally permitting CD4 T cell activation through the normal route.
5.2.7 Subversion of Antigen Processing by Pathogens
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


