An unusual disease, mastocytosis challenges the pathologist with a variety of morphologic appearances and heterogeneous clinical presentations ranging from skin manifestations (pruritus, urticaria, dermatographism) to systemic signs and symptoms indicative of mast cell mediator release, including flushing, hypotension, headache, and anaphylaxis among others. In this article, we focus on recognizing the cytology, histopathology, clinical features, and prognostic implications of systemic mastocytosis, a clonal and neoplastic mast cell proliferation infiltrating extracutaneous organ(s) with or without skin involvement. Diagnostic pitfalls are reviewed with ancillary studies to help unmask the mast cell and exclude morphologic mimics.
Overview
Mast cell disease, or mastocytosis, includes a variety of disorders that are characterized by clonal, neoplastic proliferations of mast cells in one or multiple organs, ranging from indolent and isolated proliferations to aggressive and systemic disorders. Mastocytosis is now included in the myeloproliferative neoplasms category in the 2008 World Health Organization (WHO) classification in recognition of the common theme of abnormal protein tyrosine kinase function that characterizes the myeloproliferative neoplasms. The hallmark of most mastocytosis cases is the Asp816Val (D816 V) somatic mutation in the catalytic domain of the KIT gene, resulting in enhanced mast cell survival and proliferation owing to constitutive activation of the KIT tyrosine kinase activity. 2 The WHO classification of mastocytosis is as follows:
- •
Cutaneous mastocytosis
- •
Indolent systemic mastocytosis
- •
Systemic mastocytosis with associated clonal hematological non–mast cell lineage disease (SM-AHNMD)
- •
Aggressive systemic mastocytosis
- •
Mast cell leukemia
- •
Mast cell sarcoma
- •
Extracutaneous mastocytoma.
Clinical manifestations are caused by the release of chemical mediators and by infiltration of tissues by neoplastic mast cells. Morphologic detection and immunophenotypic confirmation of mast cells in tissue sections is essential for the diagnosis of mastocytosis. Mast cells can vary from collections of round cells with many fine basophilic granules to spindled forms with associated fibrosis to blastlike cells with large metachromatic granules, with the latter atypical forms correlating with the more aggressive clinical syndromes. The subtle nature of some mast cell infiltrates can be easily overlooked or masked by accompanying eosinophils, small lymphocytes, and plasma cells.
Classification of mastocytosis into systemic mastocytosis (SM) subtypes requires correlation with clinical and laboratory findings. The WHO classification of mastocytosis separates cutaneous from systemic forms and provides criteria to further subclassify the systemic forms of mastocytosis, based on the presence of specific clinical, laboratory, and pathologic findings (that are divided into “B” and “C” groups) ( Tables 1 and 2 ).
| Major | Multifocal dense infiltrates of mast cells in tissue sections b |
| Minor | >25% spindled, immature or atypical mast cells in tissue sections or bone marrow aspirate smears Detection of KIT D816 V mutation Expression of CD2 and/or CD25 in mast cells Serum total tryptase persistently exceeds 20 ng/mL c |
a 2008 World Health Organization Diagnostic Criteria for Systemic Mastocytosis.
b Infiltrate is ≥15 mast cells in aggregates in bone marrow and/or extracutaneous organs.
c Not valid if there is an associated clonal myeloid disorder.
| B Findings | |
|---|---|
| 1. Increased mast cell burden | >30% mast cell aggregates on bone marrow biopsy and/or total serum tryptase level >200 ng/mL |
| 2. Dysplasia or myeloproliferation | Hypercellular marrow, signs of myelodysplasia or abnormal myeloid proliferation, and normal or slightly abnormal blood counts, without sufficient criteria to diagnose an AHNMD |
| 3. Organomegaly | Palpable hepatomegaly without ascites or signs of liver dysfunction, palpable or radiologic lymphadenopathy (>2 cm), or palpable splenomegaly, without hypersplenism |
| C Findings | |
| 1. Cytopenias | ANC <1.0 × 10 9 /L; Hb <10 g/dL; or platelets <100 × 10 9 /L |
| 2. Liver | Palpable hepatomegaly with impaired liver function, ascites, and/or portal hypertension |
| 3. Bone | Large osteolytic lesions and/or pathologic fractures |
| 4. Spleen | Palpable splenomegaly with hypersplenism |
| 5. Gastrointestinal | Malabsorption with weight loss and/or hypoalbuminemia |
Clinical Features
Mastocytosis is clinically heterogeneous, ranging from skin lesions that spontaneously regress to aggressive malignancies with short survival. It can occur at any age with a slight male predominance. In cutaneous mastocytosis, mast cell infiltration is limited to the skin and typically presents in childhood with urticarial symptoms. It has a benign clinical course and may regress spontaneously, often around the time of puberty. In adults, cutaneous disease is more frequently associated with indolent rather than aggressive forms of SM (see the WHO classification listed previously). Thus, in adults presenting with cutaneous disease, careful staging for SM is recommended, including a physical examination, complete blood cell count, total serum tryptase, bone marrow examination, and molecular analysis for the D816 V KIT mutation; additional laboratory and radiographic studies may be indicated based on the patient’s symptoms and signs of disease. Whereas the bone marrow is almost always involved in SM, the spleen, lymph nodes, liver and gastrointestinal tract, and virtually any organ, can also be affected. Clinical manifestations of SM reflect either mediator release from mast cells or infiltration of mast cells into tissues; they include constitutional signs, skin lesions, mediator-related findings (flushing, syncope, diarrhea, hypotension, headache, and/or abdominal pain), and musculoskeletal disease.
Four major types of SM are known :
- 1.
Indolent systemic mastocytosis
- 2.
Systemic mastocytosis accompanied by an associated hematological non–mast cell disorder (SM-AHNMD)
- 3.
Aggressive systemic mastocytosis and variant lymphadenopathic mastocytosis with eosinophilia
- 4.
Mast cell leukemia.
Indolent systemic mastocytosis involves skin and bone marrow and is the most common form of SM. Variants of indolent SM include bone marrow mastocytosis, in which no skin disease is present, smoldering systemic mastocytosis in which 2 or more “B findings” are present (see Table 2 ), and well-differentiated (round cell) mastocytosis , discussed later. Smoldering SM mainly affects older patients and is associated with more constitutional symptoms than the other types of indolent disease.
In systemic mastocytosis accompanied by an associated hematological non–mast cell disorder (SM-AHNMD), the associated non–mast cell disorder is usually a myeloid malignancy, but may also include lymphomas or plasma cell neoplasms. Symptoms and prognosis typically reflect the associated non–mast cell disease.
Aggressive systemic mastocytosis is a disorder typically lacking skin lesions and presenting with one or more “C findings” that indicate organ dysfunction owing to mast cell infiltration (see Table 2 ). A variant of aggressive SM is lymphadenopathic mastocytosis with eosinophilia, which presents with lymphadenopathy and eosinophilia. This subtype should be differentiated from myeloid/lymphoid neoplasms with PDGFRA rearrangements.
The last major type is the rare mast cell leukemia (comprising only 1% of SM cases), which also presents without skin lesions and shares the extremely poor prognosis of aggressive SM. Two localized extracutaneous mast cell neoplasms, mast cell sarcoma and extracutaneous mastocytoma, are exceedingly rare and are not discussed in this article.
Microscopic Features
Mast Cell Cytology
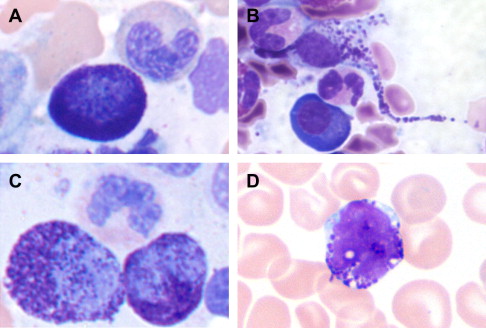
The spectrum of reactive and neoplastic mast cell appearances is presented in Table 3 . Reactive tissue mast cells are small with round, centrally located nuclei, indistinct to absent nucleoli, and abundant cytoplasm containing faint cytoplasmic granules on hematoxylin and eosin (H&E)-stained tissue sections. In Romanowsky-stained smears, normal mast cells contain tightly packed uniform metachromatic granules with round to oval-shaped nuclei ( Fig. 1 ). Neoplastic mast cell morphology has been classified into 3 subtypes (see Fig. 1 ) that are best recognized on bone marrow aspirate smears. The first subtype is the atypical mast cell type I , a spindled mast cell with elongated cytoplasmic projections, oval eccentric nuclei, and hypogranulated cytoplasm. The second subtype is the atypical mast cell type II or promastocyte , with bilobed or polylobed nuclei and typically less cytoplasmic granulation than reactive mast cells. The third subtype is the metachromatic blast, which has a high nuclear to cytoplasmic ratio, fine nuclear chromatin, prominent nucleoli, and several metachromatic granules. Atypical type I mast cells are more commonly seen in indolent types of SM, whereas atypical type II mast cells and metachromatic blasts are more common in patients with mast cell leukemia and myelomastocytic leukemia; these latter disorders are associated with a poor prognosis and short survival.
| Mast Cell Type | Size/Shape | Nucleus | Nuclear Chromatin | Cytoplasm | Nuclear to Cytoplasmic Ratio | Associated Disorders |
|---|---|---|---|---|---|---|
| Reactive | Small-medium, round or oval | Central, round or oval | Condensed | Well granulated, may be hypo- or degranulated | Low | Reactive mastocytosis |
| Atypical type I | Elongated cytoplasmic extensions (spindle shaped) | Central or eccentric, oval | Condensed | Hypogranulated, focal granule accumulation without degranulation | Variable | SM, SM-AHNMD |
| Atypical type II | Variable | Bi- or polylobed | Fine or condensed | Hypogranulated without degranulation | Variable | MCL, MML |
| Metachromatic a blast | Medium-large, round or oval | Prominent nucleoli | Fine | Few metachromatic granules | High | MCL, MML |
a Metachromatic refers to a cell that characteristically takes on a color different from that of the dye with which it is stained; a metachromatic blast has the nuclear features of a blast with many large basophilic cytoplasmic granules.
Histology
In most cases, histologic evaluation of a bone marrow biopsy, coupled with immunohistochemistry and/or special stains to identify mast cells, provides the best material to diagnose SM. The diagnostic criteria for SM include examination of tissue sections for multifocal aggregates of mast cells (see Table 1 ). Five patterns of bone marrow infiltration by mast cells are described.
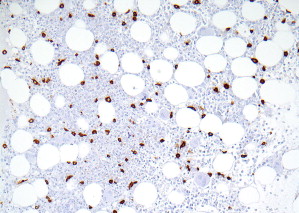
Pattern 1 is an interstitial pattern typically seen in mast cell hyperplasia ( Fig. 2 ).
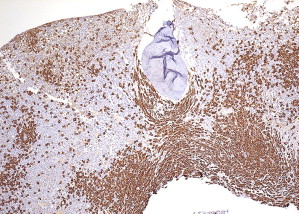
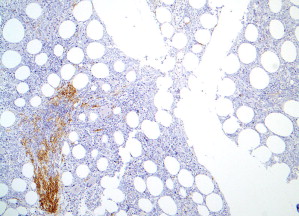
Patterns 2 through 4 involve focal, dense infiltrates of mast cells either alone (pattern 2) or coupled with additional interstitial mast cell components, located either preferentially around the focal infiltrates (pattern 3) or evenly distributed throughout the biopsy (pattern 4). Patterns 2, 3, and 4 characterize most SM cases ( Figs. 3 and 4 ).
Pattern 5 represents diffuse, dense mast cell infiltrates that obliterate the marrow architecture, and is found in mast cell leukemia and smoldering SM ( Fig. 5 ).
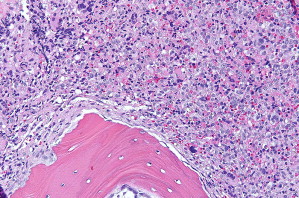
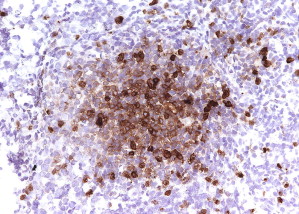
One type of focal, dense infiltrate is the tryptase-positive compact round cell infiltrate of the bone marrow (TROCI-BM) ( Fig. 6 ). This particular pattern, while rare, brings the differential diagnosis of a specific set of myeloid neoplasms, including SM, mast cell leukemia, myelomastocytic leukemia, tryptase-positive acute myeloid leukemia, acute basophilic leukemia, and SM-AHNMD ( Table 4 ).
Related posts:
 BCR-ABL Mutations in Chronic Myeloid Leukemia
BCR-ABL Mutations in Chronic Myeloid Leukemia
 Chronic Myeloid Leukemia: Mechanisms of Resistance and Treatment
Hurdles Toward a Cure for CML: The CML Stem Cell
The Biology of Chronic Myelogenous Leukemia Progression: Who, What, Where, and Why?
Portal Vein Thrombosis and Budd–Chiari Syndrome
Diagnosis of Myelodysplastic Syndromes in Cytopenic Patients
Chronic Myeloid Leukemia: Mechanisms of Resistance and Treatment
Hurdles Toward a Cure for CML: The CML Stem Cell
The Biology of Chronic Myelogenous Leukemia Progression: Who, What, Where, and Why?
Portal Vein Thrombosis and Budd–Chiari Syndrome
Diagnosis of Myelodysplastic Syndromes in Cytopenic Patients
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


