The clinical presentation of mastocytosis is diverse, and patients may not fit the classical description. Diagnosis requires a bone marrow examination, including immunohistochemical stains for mast cell tryptase or CD117. The treatment of adult systemic mastocytosis is highly individualized. This article presents an overview of systemic mastocytosis and describes its pathogenesis and treatment.
- •
The clinical presentation of mastocytosis is diverse, and patients may not fit the classical description.
- •
Diagnosis requires a bone marrow examination, including immunohistochemical stains for mast cell tryptase or CD117.
- •
The treatment of adult systemic mastocytosis is highly individualized.
Disease overview, classification, and diagnosis
The discovery of mast cells in 1878 is credited to Paul Ehrlich, who first described cells that stained with metachromatic dyes, such as toluidine blue, which he termed mast cells (MCs). MCs are myeloid lineage cells that arise from bone marrow (BM) precursors, more specifically from CD34+ and KIT+ hematopoietic progenitors. Mastocytosis is a stem cell–derived clonal myeloproliferation characterized by the abnormal growth and accumulation of neoplastic MCs in one or more organs; it is 1 of 8 subcategories of myeloproliferative neoplasms (MPNs) per the 2008 World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissues. The clinical presentation of mastocytosis is diverse, and patients may not fit the classical description; namely, a variably long history of urticaria pigmentosa (UP), followed by the insidious onset of flushing, cramping abdominal pain, diarrhea, and bone pain. Other disease manifestations include osteopenia, hepatosplenomegaly, and abnormalities of blood and BM. Unlike pediatric cases, most adults with UP-like skin lesions have systemic disease (ie, systemic mastocytosis [SM]) at presentation, a condition generally confirmed by means of a BM biopsy. The 2008 WHO system classifies mastocytosis into the following categories ( Box 1 ): (1) cutaneous mastocytosis (limited to the skin; variants include UP, diffuse cutaneous mastocytosis, and solitary mastocytoma of the skin), (2) extracutaneous mastocytoma (unifocal nondestructive MC tumor with low-grade cellular atypia), (3) MC sarcoma (destructive unifocal MC tumor with poorly differentiated MCs, and tendency to metastasize and/or evolve into MCL), and (4) SM. The latter is subclassified into 4 subcategories: indolent SM (ISM; no evidence of extracutaneous organ dysfunction), aggressive SM (ASM; presence of extracutaneous organ dysfunction), SM associated with another clonal hematological non-MC lineage disease (SM-AHNMD), and MC leukemia (MCL).
- 1.
Cutaneous mastocytosis (CM):
- a.
Urticaria pigmentosa (UP)/Maculopapular CM
- b.
Diffuse CM
- c.
Solitary mastocytoma of skin
- a.
- 2.
Indolent systemic mastocytosis
- a.
Smoldering SM
- b.
Isolated BM mastocytosis
- a.
- 3.
Systemic mastocytosis with an associated clonal hematological non-MC lineage disease (SM-AHNMD)
- 4.
Aggressive systemic mastocytosis (ASM)
- a.
Lymphadenopathic mastocytosis with eosinophilia
- a.
- 5.
Mast cell leukemia (MCL)
- 6.
Mast cell sarcoma (MCS)
- 7.
Extracutaneous mastocytoma
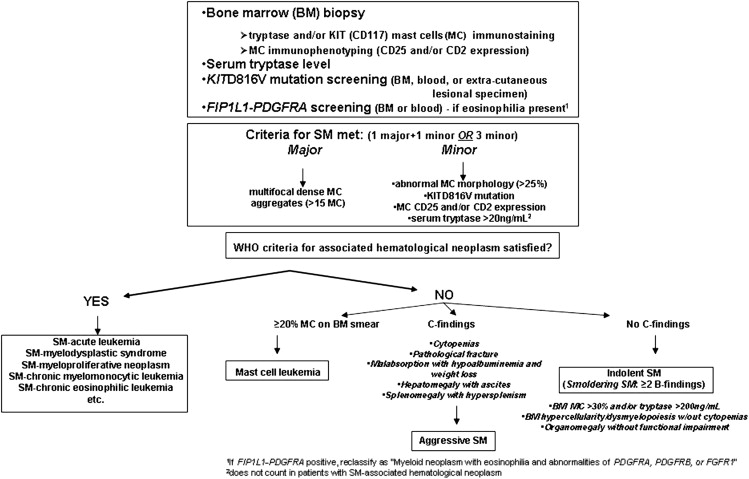
Diagnosis requires a BM examination, including immunohistochemical stains for MC tryptase or CD117 (KIT receptor) ( Box 2 , Fig. 1 ). Histologic diagnostic criteria for non-BM, extracutaneous organ involvement in SM have not been established as of yet. MCs may not be readily recognized by standard dyes, such as Giemsa, particularly when MCs exhibit significant hypogranulation or abnormal nuclear morphology. In contrast, tryptase is a sensitive marker and identifies virtually all MCs regardless of maturation or activation stage, or tissue of localization; however, neither tryptase nor CD117 immunostaining are able to distinguish between normal and neoplastic MCs. Neoplastic MCs generally express CD25 and/or CD2 (particularly the former), and detection of abnormal expression of at least 1 of the 2 antigens counts as a minor diagnostic criterion per the WHO system (see Box 2 ). Consequently, immunostaining studies enhance the morphologic and immunophenotypic distinction between normal (round and CD25-negative) and abnormal (spindle-shaped and CD25-positive) MCs, respectively. The pathognomonic BM histologic finding is presence of multifocal, dense MC aggregates, frequently in perivascular and/or paratrabecular locations. These aggregates may be relatively monomorphic, composed mainly of fusiform MCs, or may be polymorphic with MCs admixed with lymphocytes, eosinophils, neutrophils, histiocytes, endothelial cells, and fibroblasts. Eosinophils are most commonly found distributed at the periphery of MC aggregates, but increased eosinophils may also be seen in noninfiltrated areas. Identification of the KIT D816V mutation and elevated serum tryptase level (>20 ng/mL) count as a minor diagnostic criteria per the WHO system (see Box 2 ). Elevated serum tryptase levels have been documented in patients with non-SM myeloid malignancies, including acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), and chronic myeloid leukemia (CML) ; thus, non-SM myeloid disorders need to be excluded before elevated serum tryptase levels can be considered in support of a diagnosis of SM. Furthermore, levels of total serum tryptase may also be transiently elevated during anaphylaxis or a severe allergic reaction.
Major
- a.
Multifocal, dense infiltrates of MCs (≥15 MCs in aggregates) detected in sections of bone marrow and/or other extracutaneous organs
Minor
- a.
In biopsy sections of BM or other extracutaneous organs, more than 25% of the MCs in the infiltrate are spindle shaped or have atypical morphology or, of all MCs in BM aspirate smears, more than 25% are immature or atypical
- b.
Detection of an activating point mutation at codon 816 of KIT in BM, blood, or other extracutaneous organ
- c.
MCs in BM, blood, or other extracutaneous organ express CD2 and/or CD25 in addition to normal MC markers
- d.
Serum total tryptase persistently exceeds 20 ng/mL (unless there is an associated clonal myeloid disorder, in which case this parameter is not valid)
Pathogenesis
MCs retain surface KIT expression at high levels on maturation, and the interaction between the KIT and SCF has been shown to promote the proliferation, maturation, adhesion, chemotaxis, and survival of MCs. Consequently, gain-of-function mutations in KIT , particularly the D816V mutation, have been found to occur in most adult patients with SM, irrespective of WHO SM subtype. Other less common (<5%) somatic KIT mutations identified in adult SM include V560G, D815K, D816Y, insVI815-816, D816F, D816H, and D820G. Recent studies have suggested that pediatric mastocytosis is also clonal in nature, and is associated with germline or acquired activating KIT mutations. In one study that screened for KIT mutations in skin lesional DNA, 42% of cases harbored missense mutations targeting KIT D816; in 44% of cases, heterogeneous genetic alterations were found to mainly involve exons 8 and 9, which encode the fifth immunoglobulin (D5) domain and the extracellular region near the transmembrane domain. Similar to KIT D816V, every mutation in exons 8 and 9 that was functionally interrogated was found to constitutively activate KIT kinase activity. Other rare germline KIT mutations that target the transmembrane domain and that are associated with familial mastocytosis include F522C and A533D.
The issue as to whether other genetic events, in addition to activating KIT mutations, are necessary for neoplastic transformation of MCs, and for full expression of the mastocytosis phenotype, remains to be clarified. This is illustrated by the divergent natural history of childhood-onset and adult-onset mastocytosis; although both are associated with activating KIT mutations, the former predominantly exhibits cutaneous-limited disease that spontaneously regresses with age In contrast, adult-onset mastocytosis is characterized by a persistent multiorgan involvement, often with a concurrent non-MC hematologic neoplasm. Experimental data from murine models have not been conclusive in this regard; in transgenic mice expressing human KIT D816V in mature MCs (driven by the chymase promoter), only a subset (30%) of mice developed a limited form of mastocytosis (some with cutaneous-limited disease) at an older age (12–18 months). The incomplete disease penetrance in this model suggested that additional somatic mutations are necessary for full MC transformation, or that processing and intracellular trafficking of human KITD816V is abnormal in murine cells, thereby compromising signaling by the mutant receptor in this cross-species setting. In another transgenic mouse model, conditional expression of murine KITD814V was driven by the KIT promoter. When mutant KIT was expressed in somatic cells of adult mice, the animals developed marked mastocytosis with 100% penetrance at a young age, with infiltrates of mast cells in the skin and other organs, most frequently lymph nodes and colon. This phenotype reproduced some of the features of human aggressive SM and approximately half of the mice developed a non-MC lineage hematologic neoplasm, most frequently a leukemic disease derived from an immature B-cell precursor. The severity of mast cell disease was markedly attenuated when KITD814V expression in this model was limited to more mature MCs. Although half of the mice developed MC tumors, the disease occurred significantly later and progressed much slower; there is concern, however, that the attenuated phenotype may reflect low transgene expression in this model. Cumulatively, the data suggest that the effects of constitutive KIT signaling depend on the developmental stage of the cell targeted by the gain-of-function mutation. As has been noted in patients with mastocytosis, mutations targeting undifferentiated progenitors result in multilineage involvement and expression of a severe systemic disease phenotype; in contrast, mutations that target committed MC progenitors or mature MCs result in milder forms of the disease.
Recently, a high level of expression of microphthalmia-associated transcription factor (MITF), a transcription factor critical for MC development, was demonstrated in BM biopsy specimens from SM patients. Upregulation of MITF expression was found to be dependent on KIT signaling in both normal and malignant MCs. Because MITF mRNA levels did not change significantly with KIT signaling, it suggested posttranscriptional regulation of MITF expression. An array screen from MC identified miR-539 and miR-381 to be downregulated by KIT signaling; the 2 miRNAs were shown to repress MITF expression through conserved binding sites in the MITF 3′-untranslated region. These data suggest that MITF overexpression may be an important contributor to cell proliferation in mastocytosis and illustrate the role of additional genetic defects that may cooperate with KIT mutations in contributing to the fully transformed MC phenotype. It should be pointed out, however, that high MITF expression is not found in all cases of mastocytosis and the critical gene targets of MITF implicated in MC proliferation remain to be identified.
TET2 (TET oncogene family member 2) is a putative tumor suppressor gene located at chromosome 4q24. TET2 mutations were first described in patients with JAK2 V617F-positive MPN. Subsequently, the same mutations were found in JAK2 V617F-negative MPN. TET2 mutations have since been described in other myeloid malignancies, including SM, MDS, and AML. In one study, TET2 mutation frequency in SM was 29% and TET2 mutations were associated with monocytosis. In this preliminary analysis, TET2 mutations cosegregated with KIT D816V but did not appear to have an independent impact on survival. Overall, the pathogenetic role and/or prognostic impact of TET2 mutations in SM currently remain unclear.
Pathogenesis
MCs retain surface KIT expression at high levels on maturation, and the interaction between the KIT and SCF has been shown to promote the proliferation, maturation, adhesion, chemotaxis, and survival of MCs. Consequently, gain-of-function mutations in KIT , particularly the D816V mutation, have been found to occur in most adult patients with SM, irrespective of WHO SM subtype. Other less common (<5%) somatic KIT mutations identified in adult SM include V560G, D815K, D816Y, insVI815-816, D816F, D816H, and D820G. Recent studies have suggested that pediatric mastocytosis is also clonal in nature, and is associated with germline or acquired activating KIT mutations. In one study that screened for KIT mutations in skin lesional DNA, 42% of cases harbored missense mutations targeting KIT D816; in 44% of cases, heterogeneous genetic alterations were found to mainly involve exons 8 and 9, which encode the fifth immunoglobulin (D5) domain and the extracellular region near the transmembrane domain. Similar to KIT D816V, every mutation in exons 8 and 9 that was functionally interrogated was found to constitutively activate KIT kinase activity. Other rare germline KIT mutations that target the transmembrane domain and that are associated with familial mastocytosis include F522C and A533D.
The issue as to whether other genetic events, in addition to activating KIT mutations, are necessary for neoplastic transformation of MCs, and for full expression of the mastocytosis phenotype, remains to be clarified. This is illustrated by the divergent natural history of childhood-onset and adult-onset mastocytosis; although both are associated with activating KIT mutations, the former predominantly exhibits cutaneous-limited disease that spontaneously regresses with age In contrast, adult-onset mastocytosis is characterized by a persistent multiorgan involvement, often with a concurrent non-MC hematologic neoplasm. Experimental data from murine models have not been conclusive in this regard; in transgenic mice expressing human KIT D816V in mature MCs (driven by the chymase promoter), only a subset (30%) of mice developed a limited form of mastocytosis (some with cutaneous-limited disease) at an older age (12–18 months). The incomplete disease penetrance in this model suggested that additional somatic mutations are necessary for full MC transformation, or that processing and intracellular trafficking of human KITD816V is abnormal in murine cells, thereby compromising signaling by the mutant receptor in this cross-species setting. In another transgenic mouse model, conditional expression of murine KITD814V was driven by the KIT promoter. When mutant KIT was expressed in somatic cells of adult mice, the animals developed marked mastocytosis with 100% penetrance at a young age, with infiltrates of mast cells in the skin and other organs, most frequently lymph nodes and colon. This phenotype reproduced some of the features of human aggressive SM and approximately half of the mice developed a non-MC lineage hematologic neoplasm, most frequently a leukemic disease derived from an immature B-cell precursor. The severity of mast cell disease was markedly attenuated when KITD814V expression in this model was limited to more mature MCs. Although half of the mice developed MC tumors, the disease occurred significantly later and progressed much slower; there is concern, however, that the attenuated phenotype may reflect low transgene expression in this model. Cumulatively, the data suggest that the effects of constitutive KIT signaling depend on the developmental stage of the cell targeted by the gain-of-function mutation. As has been noted in patients with mastocytosis, mutations targeting undifferentiated progenitors result in multilineage involvement and expression of a severe systemic disease phenotype; in contrast, mutations that target committed MC progenitors or mature MCs result in milder forms of the disease.
Recently, a high level of expression of microphthalmia-associated transcription factor (MITF), a transcription factor critical for MC development, was demonstrated in BM biopsy specimens from SM patients. Upregulation of MITF expression was found to be dependent on KIT signaling in both normal and malignant MCs. Because MITF mRNA levels did not change significantly with KIT signaling, it suggested posttranscriptional regulation of MITF expression. An array screen from MC identified miR-539 and miR-381 to be downregulated by KIT signaling; the 2 miRNAs were shown to repress MITF expression through conserved binding sites in the MITF 3′-untranslated region. These data suggest that MITF overexpression may be an important contributor to cell proliferation in mastocytosis and illustrate the role of additional genetic defects that may cooperate with KIT mutations in contributing to the fully transformed MC phenotype. It should be pointed out, however, that high MITF expression is not found in all cases of mastocytosis and the critical gene targets of MITF implicated in MC proliferation remain to be identified.
TET2 (TET oncogene family member 2) is a putative tumor suppressor gene located at chromosome 4q24. TET2 mutations were first described in patients with JAK2 V617F-positive MPN. Subsequently, the same mutations were found in JAK2 V617F-negative MPN. TET2 mutations have since been described in other myeloid malignancies, including SM, MDS, and AML. In one study, TET2 mutation frequency in SM was 29% and TET2 mutations were associated with monocytosis. In this preliminary analysis, TET2 mutations cosegregated with KIT D816V but did not appear to have an independent impact on survival. Overall, the pathogenetic role and/or prognostic impact of TET2 mutations in SM currently remain unclear.
Related posts:
 Disordered Signaling in Myeloproliferative Neoplasms
Disordered Signaling in Myeloproliferative Neoplasms
 Genotype-Phenotype Interactions in the Myeloproliferative Neoplasms
Genotype-Phenotype Interactions in the Myeloproliferative Neoplasms
 Clinical Predictors of Outcome in MPN
Acquired Uniparental Disomy in Myeloproliferative Neoplasms
Myeloproliferative Neoplasm Animal Models
Clinical Predictors of Outcome in MPN
Acquired Uniparental Disomy in Myeloproliferative Neoplasms
Myeloproliferative Neoplasm Animal Models
 Therapy with JAK2 Inhibitors for Myeloproliferative Neoplasms
Therapy with JAK2 Inhibitors for Myeloproliferative Neoplasms
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


