Abstract
Thyroid hormone (TH) synthesis and release is under feedback regulation by the hypothalamic input to the pituitary via thyroid-stimulating hormone (TSH) and thyroid-releasing hormone (TRH) resulting in pituitary secretion of TSH. Clinicians generally diagnose thyroid disease based on the serum levels of the THs [ l -3,3’,5-triiodothyronine (T 3 ) and l -3,3’,5,5-tetraiodothyronine (thyroxine or T 4 )] in conjunction with the measurement of serum TSH. Patients with hypothyroidism have low serum T 4 and high serum TSH concentrations. This is usually a result of reduced production of TH due to gland destruction (such as in autoimmune thyroid disease) or impaired biosynthesis of thyroid hormone (such as in organification defects). On the other hand, hyperthyroidism is usually diagnosed when the T 4 and T 3 are high and the TSH is suppressed. The latter is commonly seen in autoimmune hyperthyroidism or autonomous TH secreting adenomas of the thyroid gland. When there is loss of the inverse relationship between TH levels and TSH concentrations (e.g., high serum T 3 and/or T 4 but normal TSH as in TH resistance) or when T 4 and T 3 levels are markedly different (e.g., high T 3 and low T 4 in a TH transporter defect, or low T 3 and high T 4 as seen in TH metabolism defects), the clinician must consider a genetic defect in the differential diagnosis. Correct diagnosis can be made from clinical observations and confirmed by appropriate genetic testing, as discussed in this chapter. The correct diagnosis will lead to a rational therapy avoiding inappropriate thyroid gland ablation or hormone supplementation.
Keywords
thyroid hormone (TH), monocarboxylate transporters (MCT), selenoproteins, hyperthyroidism, hypothyroidism, thyroid hormone receptor (THR) gene, selenocysteine insertion sequence-binding protein 2 (SBP2), laboratory findings and differential diagnosis, clinical features
Introduction
Thyroid hormone (TH) synthesis and release is under feedback regulation from the hypothalamic input to the pituitary via thyroid stimulating hormone (TSH) and thyroid-releasing hormone (TRH). Clinicians generally diagnose thyroid disease based on the serum levels of the THs [( l -3,3’,5-triiodothyronine (T 3 ) and l -3,3’,5,5’-tetraiodothyronine thyroxine, (T 4 )] in conjunction with the measurement of serum TSH. Patients with hypothyroidism have low serum THs and high TSH serum concentrations (see 7 , 8 ). This is usually a result of reduced production of TH due to gland destruction (such as in autoimmune thyroid disease) or impaired biosynthesis of TH (such as in organification defects). On the other hand, hyperthyroidism is usually diagnosed when the T 4 and T 3 are high and the TSH is low. The latter is commonly seen in autoimmune hyperthyroidism or autonomous TH secreting adenomas of the thyroid gland. When there is loss of the inverse relationship between TH levels and TSH concentrations or when T 4 and T 3 levels are markedly different (e.g., high T 3 and low T 4 in a TH transporter defect, or low T 3 and high T 4 as seen in TH metabolism defects, see further), the clinician must consider a broad differential diagnosis. Correct diagnosis can be made from clinical observations and confirmed by appropriate genetic testing, as discussed in this chapter. The correct diagnosis will lead to a rational therapy avoiding inappropriate thyroid gland ablation or hormone supplementation.
It is important to appreciate that the centrally regulated system described previously, is not affected by TH demands in cells not directly involved in the feedback control. Local requirement in TH is adjusted though additional mechanisms. One such system is the control of TH entry into the cell through active transmembrane transporters. Another is the activation of the precursor T 4 by removal of the outer ring iodine (5’-deiodination) to form T 3 or, inactivate T 4 and T 3 by inner ring (5-deiodination) to form l -3,3’,5’-triiodothyronine or reverse T 3 (rT 3 ) and l -3,3’-diiodothyronine (T 2 ), respectively ( Fig. 9.1 ). Changing concentrations of deiodinases in various cell types, allows an additional local regulation of hormone supply.
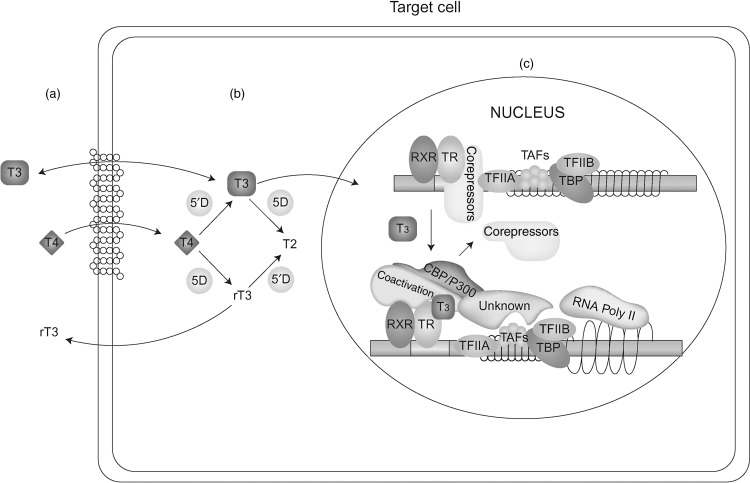
Finally, the presence and abundance of TH receptors (TRs), through which TH action is mediated, determines the type and degree of hormonal response. Action takes place in the cytosol as well as in the nucleus. The latter, known as genomic effect, has been more extensively studied ( Fig. 9.1 ). TRs are transcription factors that are associated with DNA of genes whose expression they regulate.
The syndromes of impaired sensitivity to TH include a group of disorders with apparent discordance between serum TSH and TH levels and/or hormone action at the tissue and cell level. Resistance to thyroid hormone (RTH), a syndrome of reduced end-organ responsiveness to TH was identified in 1967. With the recognition of thyroid hormone receptor beta (THRB) gene mutations the term RTH became synonymous with defects of the TR. Recent discoveries of genetic defects that reduce the effectiveness of TH through altered cell membrane transport and metabolism broadened the definition of resistance to TH to include all defects that alter the biological activity of an authentic hormone secreted in normal amounts. It is suggested that use of the acronym RTH be limited to the syndrome produced by reduced intracellular action of the active TH, T 3 . Impaired sensitivity to TH (ISTH) is used to describe altered effectiveness of TH in the broader sense. While the clinician considers these defects when confronted with thyroid function tests that show a discordance of serum TH and TSH concentrations, each defect has its own constellation of test abnormalities and different clinical presentation.
Overview of described and putative defects in syndromes of impaired sensitivity to thyroid hormone
Thyroid Hormone Cell Transport Defect (THCTD)
Altered intracellular accumulation of hormone can be caused by mutations in cell membrane hormone transport proteins ( Fig. 9.1 a). These molecules belong to different families of solute carriers, organic anions, amino acids, and monocarboxylate transporters (MCT). A defective gene may fail to synthesize a protein, form a molecule that may not reach the cell membrane, or be defective in its ability to transport the hormone. In all these instances there is lack or insufficient hormone in cells dependent on the specific hormone transporter. A defect in one such transporter, MCT8, presents elevated serum concentrations of T 3 and low levels of T 4 and rT 3 accompanied by severe psychomotor deficit.
Thyroid Hormone Metabolism Defect (THMD)
T 4 , the major product secreted by the thyroid gland, is a prohormone requiring activation through its conversion to T 3 ( Fig. 9.1 b). Defects in any of the factors involved in this enzymatic reaction can cause a diminished production of T 3 and thus, reduced sensitivity to the hormone. Defects may include abnormalities in the synthesis or degradation of the various deiodinases. One such defect has been recently identified reducing the synthesis of selenoproteins, a family of proteins to which deiodinases belong. Patients present low serum T 3 and high T 4 and rT 3 concentrations.
Abnormal Hormone Transfer to the Nucleus
The main and best-studied TH effect requires the translocation of the hormone into the nucleus where it interacts with the TR ( Fig. 9.1 c). Putative defects in the transfer of hormone and/or its receptor to the nucleus are expected to reduce the hormonal action at the genomic level. No such defects have been yet described.
Thyroid Hormone Receptor Defects Resulting in RTH
Nuclear, also known as genomic action of TH is mediated through the TRs ( Fig. 9.1 c). Mutant TR proteins have reduced ability to bind cognate ligand or protein cofactors and possibly DNA. Mutations have been identified in each of the two THR genes, alpha and beta. Patients with thyroid hormone receptor beta (THRB) gene mutations have RTHβ that manifests persistent elevations of all three serum iodothyronines with nonsuppressed TSH. Clinical manifestation are usually minor and could suggest both hypo- and hyperthyroidism. Those with mutations in the THRA gene have subtle abnormalities in serum thyroid tests as TRα has a minor role in the feedback regulation of thyroid hormone synthesis and secretion. The cardinal features of sort stature, constipation and intellectual impairment reflect the organs that are TRα dependent.
Abnormal Cofactors or Interfering Substances
Nuclear hormone receptors exert the hormone-mediated activity by forming a complex that involves accessory and modulatory proteins as well as the ligand ( Fig. 9.1 c). Putative defects in cofactors that normally stabilize the hormone-receptor complex or cofactors that repress or activate function could be responsible for reduced hormone sensitivity. Patients without THRB gene mutations but presenting a phenotype indistinguishable from that manifested in the presence of a mutation, are believed to belong to this category of abnormalities.
“Nongenomic” Abnormalities
Recent work has established that TH can also act at the level of the cytosol through nongenomic mechanisms. It is anticipated that impairment of such activity would result in yet unrecognized disease states.
Resistance to thyroid hormone (RTH)
Background
With the recent discovery of TRα mutations in humans and their distinct phenotype, it has been proposed to classify the RTH syndromes based on their molecular etiology, namely: (1) RTHβ for classic RTH due to mutations in the THRB gene; (2) NonTR-RTH for patients with an identical phenotype as RTHβ but lacking THRB gene mutations; (3) RTHα for those subjects with a distinct phenotype harboring THRA gene mutations.
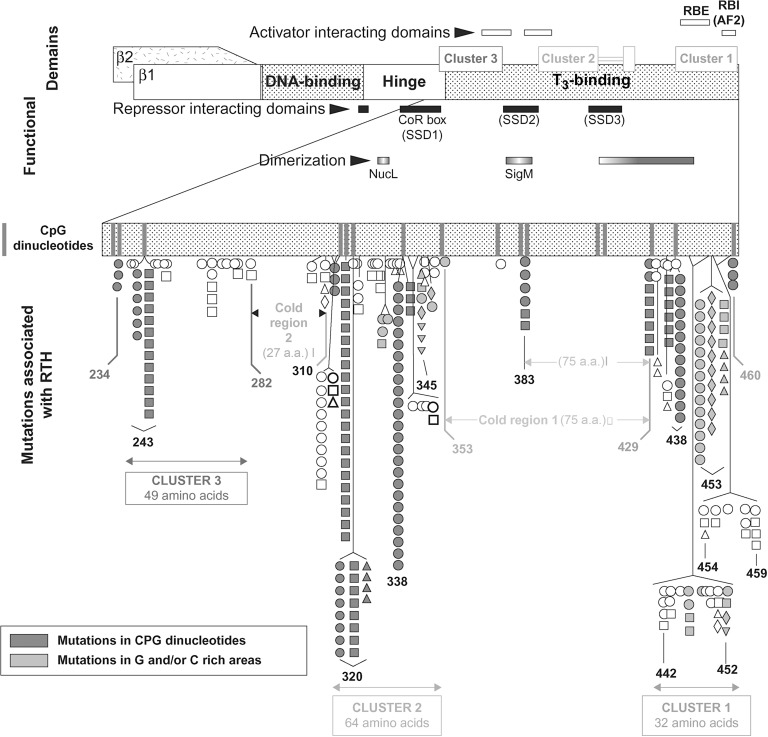
Expression of TH effects requires the presence in the cell of sufficient amount of the active hormone, T 3 . The best-studied effect involves the translocation of T 3 into the nucleus where it interacts with TRs to regulate (activate or repress) transcription of target genes. There are two TRs, α and β, which are encoded by separate genes located on chromosomes 17 and 3, respectively. The receptors have structural and sequence similarities and exist as different isoforms with DNA-binding and T 3 -binding domains ( Fig. 9.2 ). Other regions of the molecules are involved in dimerization with another TR (homodimerization) or another type of nuclear receptor (heterodimerization), and in binding corepressor and coactivator protein cofactors ( Fig. 9.1 c).
Dimers of unliganded (without T 3 ) receptors bind to TH-response elements(TRE), resulting in inhibition of the expression of genes that are positively regulated by T 3 through association with corepressor proteins. T 3 -binding to the receptors produces a stearic change of the TR molecule that results in release of the corepressor protein, dissociation of the dimers, and formation of heterodimers of TR and retinoid X receptors that then bind coactivator proteins ( Fig. 9.1 c). These changes promote gene expression and ultimately increase the synthesis of specific proteins.
RTHβ
RTHβ is an inherited syndrome characterized by reduced responsiveness of target tissues to TH. It is first suspected by the findings of high serum concentrations of free T 4 and usually also free T 3 , accompanied by normal or slightly high serum TSH concentration.
Incidence and Prevalence
RTH has been detected in 1 of 40,000 live births, and it occurs with equal frequency in both sexes. With the exception of a single family, it is inherited as an autosomal dominant trait. In the past, RTH was subdivided into generalized, isolated pituitary and isolated peripheral tissues resistance. This classification, based upon clinical findings alone, has no genetic basis as the former two can occur in individuals with the same mutation. The latter represents the development of tolerance to the ingestion of excess TH.
Current published cases surpass 3000 and of the 460 families with RTH, 50% were investigated in the authors’ institution. Of the latter, 34 families (15%) had no THRB gene mutations and 48 (21 %) of the mutations occurred de novo . The latter commonly occur in CpG dinucleotide hot spots or in guanine or cytosine reach areas ( Fig. 9.2 ).
Clinical Features
The hallmark of RTHβ is the paucity of symptoms and signs of thyroid dysfunction despite the presence of high serum T 4 and T 3 concentrations. Affected individuals may have variable symptoms or signs of hypothyroidism or hyperthyroidism.
Growth retardation, delayed bone maturation, learning disabilities, mental retardation, sensorineural deafness, and nystagmus are compatible with hypothyroidism, while tachycardia, hyperactivity, and increased basal metabolic rate are suggestive of hyperthyroidism. Overt hypothyroidism is more common in those patients who, because of erroneous diagnosis of hyperthyroidism, received treatment to decrease thyroid secretion. There is a high prevalence of attention deficit hyperactivity disorder and learning disabilities. Hearing loss may be due in part to recurrent ear infections, which are more common in the patients with RTH, but sensorineural deafness is typical of RTH due to TRβ gene deletion in humans. In mice, absence of TRβ1 is sufficient to cause hearing loss.
Among all clinical findings, goiter is by far the most common (65–95 %), followed by hyperactivity (33–68%), and tachycardia (33–75 %). It is these abnormalities that usually lead to evaluation of thyroid function. The subsequent finding of high serum T 4 and T 3 concentrations often results in the erroneous diagnosis of hyperthyroidism.
Laboratory Findings and Differential Diagnosis
In the untreated subject, a high serum free T 4 concentration and nonsuppressed TSH are sine qua non requirements for the diagnosis of RTHβ. Serum levels of T 3 and rT 3 are usually also high. Thyroglobulin concentration tends to be high, reflecting the level of TSH induced thyroid gland hyperactivity. The response of TSH to TSH-releasing hormone is normal or exaggerated, depending on the baseline TSH level. The suppressive effect of administered TH on TSH, cholesterol, and creatine kinase, and the stimulatory effect on sex-hormone binding globulin and ferritin are attenuated.
All causes of high serum T 4 and T 3 concentrations in association with normal to high serum TSH levels should be considered in the differential diagnosis, after the abnormalities are confirmed by repeat measurements several weeks later ( Table 9.1 ). The next step is to measure serum free T 4 and T 3 , preferably by equilibrium dialysis; normal values suggest a defect of TH transport in serum, not RTH.
| Defect | Thyroid function tests | Prevalence | |||||
|---|---|---|---|---|---|---|---|
| T 4 | T 3 | rT 3 | TSH | FT 4 direct | FT 4 dialysis | ||
| Abnormal binding protein | |||||||
| ↑TBG | ↑ | ↑ | ↑ | N | N | N | 1:100 males |
| ↑TTR * | ↑ | N | ↑ | N | N | N | 1:10,000 |
| FDH | ↑ | ↑ or N | ↑ | N | ↑↑ | N | 1:600 |
| Reduced sensitivity to TH | |||||||
| RTHβ | ↑ | ↑ or N | ↑ | sl↑ or N | ↑ | ↑ | 1:40,000 |
| TSHoma ** | ↑ | ↑ | ↑ | sl↑ or N | ↑ | ↑ | Unknown |
| THCTD | ↓ | ↑↑ | ↓↓ | sl↑ or N | ↑ | ↑ | Unknown 108 families |
| THMD | ↑↑ | ↓ | ↑↑ | ↑ or N | ↑↑ | ↑↑ | Unknown 9 families |
| RTHα | ↓ | ↑ or N | ↓↓ | sl↑ or N | ↓ | ↓ | Unknown 5 families |
* Refers to TTR with increased affinity for T 4 and rT 3.
** To distinguish between RTH and TSHoma please see text. In brief: response to TRH stimulation, lower TSH value, lower alpha subunit, family history, and presence of mutation are consistent with RTH.
The demonstration of similar abnormalities in serum T 4 and TSH concentrations in first-degree relatives obviates the need to exclude a TSH-producing pituitary adenoma (TSHoma). A high ratio of the alpha subunit to whole TSH is pathognomonic of TSHoma. The latter disorder is associated with a similar TH profile as RTH but most, if not all, patients are hyperthyroid.
The ability to identify mutations in the THRB gene provides a means to confirm the diagnosis, to obtain prenatal diagnosis, and to prevent inappropriate antithyroid treatment of patients with high serum levels of free TH.
Genetic Pathophysiology
RTHβ is caused by mutations in the THRB gene, located on chromosome 3. Tissues in patients with RTHβ are resistant to action of T 3 to the extent this gene is expressed in the cells involved. In the majority of cases, mutations have been found in the carboxyl terminus of the TRβ covering the ligand-binding domain and adjacent hinge domain ( Fig. 9.2 ). They are contained within three clusters rich in CpG “hot spots”, separated by areas devoid of mutations (cold regions). The latter are located between codons 282 and 310, and with the exception of 383, codons 353 and 429. No mutation has been reported upstream of codon 234. As cold regions are not devoid of “hot spots,” the lack of mutations reflects the observation that mutations in the second cold region do not impair TR function and therefore, are not expected to produce a phenotype.
THRB gene defects have been identified in 426 families comprising 132 distinct mutations. The authors have found mutations in 213 families and a partial listing is available from http://www.receptors.org/cgi-bin/nrmd/nrmd.py . Although mostly are missense mutations, nucleotide deletion, and insertions producing frameshifts have created in five instances nonsense proteins with two additional aminoacids. In four instances single nucleotide deletions have produced truncated receptors. In only one family complete THRB gene deletion resulted in recessively inherited RTH. The following mutations have been identified in more than 10 unrelated families, often the consequence of de novo mutations: R243Q (15 families); A317T (29 families); R338W (30 families); R438H (17 families); P453T (17 families); and P453S (12 families).
The mutant TRβ molecules have either reduced affinity for T 3 or impaired interaction with one of the cofactors involved in the mediation of TH action. As TR mutants are still able to bind to TREs on DNA and dimerize with normal TRs or the RXR partner, they interfere with the function of the normal TRs, explaining the dominant mode of inheritance. Therefore, it is not surprising that in the single family reported with a deletion of all coding sequences of the THRB gene, only homozygotes manifest the phenotype of RTHβ⋅
A family with two de novo THRB gene mutations occurring in the same nucleotide has been reported. The proposita with apparent de novo missense mutation (GTG to GGG) in codon 458 of the THRB gene (V458G), transmitted this mutation to her affected son. The mutant allele underwent another de novo mutation transferred to her affected daughter as GAG (V458E). This apparent attempt of repair is more likely the result of the creation of a mutagenic prone three guanines sequence by the first mutation.
A poorly understood feature of RTH is the variable severity of hormonal resistance in different tissues in the same patient and among different patients having the same mutation. Studies of transgenic mice bearing Thrb gene mutations and gene deletions have provided some insight into these differences. As an example, the tachycardia that occurs in patients with RTHβ is due to the unopposed activation of the TRα by the high serum T 4 and T 3 concentrations in the heart that expresses predominantly this TR isoform. The variable manifestations of RTH among affected sibs with the same mutation is unexplained, but they likely result from genetic variability of cofactors involved in TH action.
Treatment
There is no treatment that will correct the defect of TRβ function in subjects with RTHβ. Fortunately, in most patients, the hyposensitivity to TH seems to be adequately compensated by the increase in secretion of T 4 and generation of T 3 . Thus, treatment is usually not needed. This is not the case in patients with limited thyroid reserve due to prior destructive therapy directed to the thyroid gland. These patients should be treated with sufficient amount of levothyroxine ( l -T 4 ) to reduce their serum TSH concentrations to normal or near normal.
In some patients with RTHβ, several peripheral tissues may be relatively more resistant than the thyrotrophs. Thus, the compensation for the hormonal resistance in these tissues is incomplete and judicious administration of a dose of l -T 4 higher than that needed to restore TSH secretion to normal may be indicated. The dose must be individually determined by assessing tissue responses. In children, this should be done by regular assessment of growth, bone maturation, and mental development. l -T 4 should be given in incremental doses, and the basal metabolic rate, nitrogen balance, and serum sex hormone-binding globulin should be measured after treatment for 4–6 weeks before the dose is changed; bone age and growth should be followed on a longer term basis. Development of a catabolic state is an indication of overtreatment.
Management of TH levels during pregnancy in a mother with RTHβ or a normal mother carrying a fetus with RTHβ is not straightforward. A retrospective study of a large family with RTHβ demonstrated that the adverse effect of TH on the fetus was independent of that on the pregnant woman who, because of the resistance, is protected from the metabolic effect of the hormone.
The prevalence of early pregnancy loss was increased fivefold in affected mothers, but not in couples with an affected father and unaffected mother. Two-thirds of their infants carried the THRB gene mutation, which suggests that nearly all miscarried fetuses had no mutation and thus, a normal response to TH. Furthermore, unaffected infants born to affected mothers had lower birth weights and suppressed serum TSH concentrations when compared to affected infants. These results are in agreement with the finding that infants with excess TH caused by gain-of-function TSH receptor gene mutations are born prematurely and have low birth weights. Therefore, management of pregnancies in mothers with RTHβ, carrying unaffected fetuses, may warrant judicious use of antithyroid medication depending on the well-being of the fetus. Further studies are needed before general recommendations can be made.
NonTR-RTH
NonTR-RTH refers to the occurrence of the RTHβ in the absence of a THRB gene mutation. It is clinically and biochemically undistinguishable from RTHβ with THRB gene mutations. In several of these families, inheritance is autosomal dominant and mutations in THRB gene have been excluded by the absence of genetic cosegregation and sequencing, thus ruling out mosaicism. Based on the observations in mice and studies in humans mutations of one of the cofactors that interact with the receptors may be responsible for the resistance in these families.
RTHα
Background, Incidence, and Prevalence
Thrα gene manipulations in the mouse have long demonstrated that there is an absence of the RTHβ phenotype and that there may be lack of central hypothyroidism and perturbations in metabolic regulations. Our knowledge of RTHα in humans is limited to only 13 subjects belonging to 9 families ( Table 9.2 ). The first case was identified by whole-exome sequencing. The proposita, a white female of European descent, was heterozygous for a de novo mutation in the THRA gene. A second family, of Greek ancestry, had an affected father and daughter. A fourth case was diagnosed at the age of 42 in a woman with epilepsy, growth retardation, constipation, and macrocephaly. Six additional families with THRA gene mutations were recently presented. The prevalence remains unknown.
| THRA gene nucleotide # | THRA protein | FT4 % lower limit of normal | FT3 % upper limit of normal | TrT3 % lower limit of normal | TSH (mU/L) | Known THRB gene mutations in corresponding codons | References |
|---|---|---|---|---|---|---|---|
| 806 C > T | A263V | 99 * | 90 * | <63 * | 4.2 * | A317V/T/S/D | |
| 1075 A > T | N359Y | 114 | 100 | 121 | 0.3 | ||
| 1144 delG | A382fsM388X | 100 | 140 | 91 | 5.8 | A436T/fs, M442V/T | |
| 1176 C > A | C392X | 107 | 148 | 2.8 | C446X/G/R | ||
| 1193 C > G | P398R | 72 | 70 | 0.5 | P452H/L/∆ | ||
| 1207 G > T | E403X | 63 | 80 | 1.0 | E457G | ||
| 1207 G > T | E403X | NA | NA | NA | E457G | ||
| 1207 G > A | E403K | 106 | 90 | 33 | 1.9 | E457G | |
| 1190 insT | F397fs E406X | Low Nl | High Nl | l | Nl | E460K |
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


