ACTH dependent
Pituitary adenoma (Cushing’s disease)
Ectopic ACTH secretion
ACTH independent
Adrenal adenoma
Adrenal carcinoma
ACTH-independent macronodular adrenal hyperplasia (AIMAH)
Primary pigmented nodular adrenal disease (PPNAD)
Other unilateral or bilateral adrenal disease
ACTH-Dependent CS
Cushing’s Disease: Pituitary Adenoma
The most common cause of ACTH-dependent CS is a pituitary adenoma or Cushing’s disease (CD) . Typically, the adenoma is relatively small in size (microadenoma) with an average diameter of < 5 mm, though larger macroadenomas (> 10 mm) do occur. It is important to note that autopsy studies suggest up to 27 % of individuals will have incidental pituitary adenomas, so the mere presence of a mass seen on imaging does not necessarily mean that the patient has CD.
Ectopic ACTH Secretion
ACTH secretion can occur from sources other than the pituitary. Small-cell lung cancer frequently gives rise to paraneoplastic syndromes and can serve as a source of excess ACTH which stimulates adrenal cortisol secretion. Additionally, most neuroendocrine tumors including pheochromocytoma, gastrinoma, and carcinoids can secrete ACTH, as can medullary thyroid cancer and neuroblastoma [4].
ACTH-Independent CS
Unilateral Disease
Unilateral adrenal nodules causing CS can be due to either adenomas (60 %) or carcinomas (40 %) with the risk of cancer increasing with size. Nodules < 4 cm carry a 2 % cancer risk, while those > 6 cm have a 25 % risk of malignancy. Intermediate-sized nodules from 4 to 6 cm are found to be cancer in 6 % of cases [5, 6].
Bilateral Disease
ACTH-Independent Macronodular Adrenal Hyperplasia
ACTH-independent macronodular adrenal hyperplasia (AIMAH) is the underlying cause in < 1 % of patients with CS and has also been referred to as massive macronodular adrenocortical disease, autonomous macronodular adrenal hyperplasia, ACTH-independent massive bilateral adrenal disease, and giant macronodular adrenal disease. Age of symptom onset is typically in the fifth or sixth decade, and men have a similar incidence as women [7, 8]. The classic imaging finding is multiple large adrenal nodules with pathology showing two distinct cell types: those containing clear cytoplasm or lipid-rich cells, and those with a compact cytoplasm or lipid-poor cells. It should be noted, however, that the disease may progress at a different rate in each adrenal gland so that what initially appears to be unilateral disease ultimately develops into bilateral nodules. Adrenal cortisol secretion is amplified due to an increased number of adrenocortical cells rather than greater production from individual cells.
Primary Pigmented Nodular Adrenocortical Disease
Unlike with AIMAH, the actual size of the adrenal gland may be normal but the structure is replaced with multiple nodules scattered throughout an atrophic cortex. On imaging, primary pigmented nodular adrenocortical disease (PPNAD) can have several different appearances ranging from a string of beads to several large nodules. On pathologic examination, most of the nodules are small (< 4 mm) and clearly separate from the surrounding cortex. The nodules seen in PPNAD generally do not show a significant response to exogenous ACTH administration but demonstrate a paradoxical increase in cortisol secretion in response to dexamethasone.
The etiology of PPNAD is evenly split between sporadic and familial causes. The Carney complex is a multiple endocrine neoplasia syndrome involving myxomas (cutaneous and cardiac), spotty skin pigmentation, schwannomas, and several endocrine tumors. PPNAD is the most common endocrine tumor seen in the Carney complex (see Chap. 37 for more information on Carney) and can be found in one fourth of those patients. Additionally, PPNAD has been linked to mutations in the cyclic adenosine monophosphate (cAMP)-dependent protein kinase A ( PRKAR1A) gene [9].
Presentation and Natural History
While the classic presentation of CS involves the characteristic physical changes such as a “buffalo hump” and purple striae, a wide array of other symptoms are associated with CS (Table 2), and the diversity of presentation frequently leads to delays in diagnosis. In particular, the manifestations of CS are quite similar to those of the metabolic syndrome and diabetes so a high index of suspicion is needed to make the diagnosis. Additional physical signs that can help distinguish CS from other disease processes include skin changes (easy bruising and fragile skin) and proximal muscle weakness.
Table 2
Signs and symptoms of Cushing’s syndrome
Sign or symptom | Percentage of patients with symptom |
|---|---|
Obesity | 79–97 |
Plethora | 78–94 |
Diabetes or impaired glucose tolerance | 39–94 |
Moon facies | 88–92 |
Hypertension | 47–90 |
Muscle weakness | 45–90 |
Menstrual changes | 35–86 |
Hirsutism | 58–84 |
Thin skin or easy bruising | 17–84 |
Bone changes | 48–83 |
Acne | 21–82 |
Depression | 25–67 |
Buffalo hump | 34–67 |
Striae | 50–64 |
CS significantly increases mortality primarily by increasing the risk of cardiovascular disease [10]. Compared to the normal population, the standardized mortality ratio for those with CS can be as high as 4.8 [11–13]. In addition to cardiovascular complications, CS is associated with hypercoagulability and an increased risk of deep vein thrombosis (DVT) and pulmonary embolus (PE) [14, 15]. This disease manifestation is particularly important when planning operative intervention. CS not only leads to physical changes and symptoms, there are also important psychological changes associated with the diagnosis. Depression and psychosis are commonly seen but more subtle abnormalities including fatigue and loss of concentration and memory also occur. Equally important is a sharp decline in measures of health-related quality of life (HRQL) covering multiple aspects of mental, social, and physical health [16–18]. In short, CS impacts multiple organ systems and the effects worsen the longer diagnosis and treatment are delayed.
Subclinical CS
A subset of patients with incidentally discovered adrenal masses will have elevated cortisol levels but lack the typical signs and symptoms associated with glucocorticoid excess. These patients are categorized as having “subclinical” CS although they often present with hypertension, obesity, hyperlipidemia, and impaired glucose tolerance . The diagnostic tests which are discussed below are suboptimal when it comes to diagnosing subclinical CS as the biochemical changes are generally subtle and erratic. Consequently, no clear consensus has emerged regarding the best criteria or approach for diagnosing the condition. The same overall approach is used but a higher level of suspicion must be maintained in order to make the diagnosis.
Diagnostic Evaluation
The diagnosis of CS is complicated by the fact that there is no widely accepted gold standard test and each of the available tests has important limitations. In general, the diagnostic process can be broken down into discrete steps beginning with confirmation of excess cortisol (Fig. 1). Once a hypercortisol state is identified, the goal is to identify whether the underlying cause is autonomous secretion of cortisol due to adrenal disease or whether ACTH (or corticotropin-releasing hormone, CRH) secretion is driving adrenal oversecretion. Finally, if the diagnosis of ACTH-dependent disease is made, it is necessary to determine whether the source of ACTH comes from the pituitary or an ectopic source.
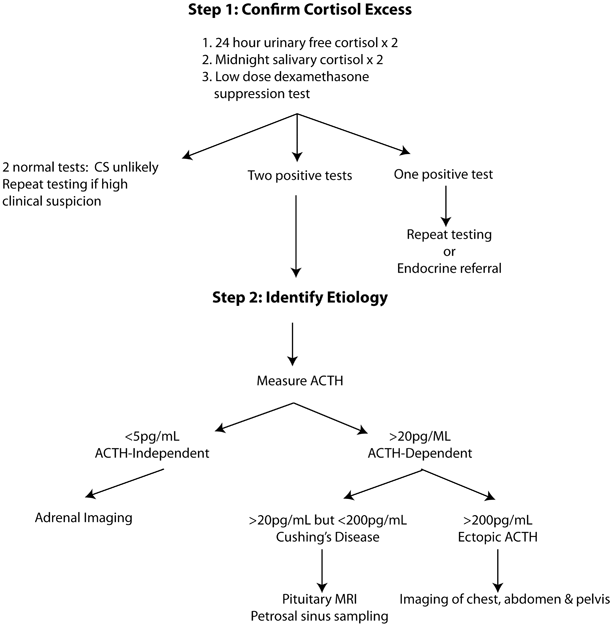
Fig. 1
Diagnostic approach to Cushing’s syndrome. ACTH adrenocorticotropic hormone, CS Cushing’s syndrome, MRI magnetic resonance imaging
Screening for Hypercortisolism
Recent guidelines from the Endocrine Society recommend the use of any of three tests for confirmation of excess cortisol secretion : low-dose dexamethasone suppression (LDDST), 24 h urine cortisol, and midnight salivary cortisol [19]. Table 3 outlines the advantages, disadvantages, and what constitute a positive result for each test. These tests were selected based on their relatively high sensitivity since the consensus opinion was that the key to the initial diagnostic step is to avoid missing those who might actually have CS. The focus on sensitivity rather than specificity was based on the significant impact that untreated CS can have on mortality. The committee felt that, on balance, it would be better to have a few false positives which could be appropriately excluded by subsequent testing rather than missing those who might have CS and benefit from rapid evaluation and treatment. Since CS may be cyclic in nature or involve subtle derangements in the hypothalamic–pituitary–adrenal axis (HPA), it is frequently necessary to conduct multiple tests to confirm the diagnosis. Since there is no ideal or gold standard test for diagnosing CS, using two or more of these screening tests in combination may offer the best hope for accurate diagnosis. Collecting two separate samples for urine-free cortisol (UFC) or salivary cortisol is also useful as concordant results add support to the diagnosis of CS while discordant results should prompt additional testing .
Table 3
Comparison of screening tests for Cushing’s syndrome
Low-dose dexamethasone suppression test | 24 h urine-free cortisol | Midnight salivary cortisol | |
|---|---|---|---|
Performance advantages | Sensitivity up to 95 % | Sensitivity > 85 %, specificity > 90 % | Sensitivity ³ 92 % |
1. Able to distinguish ACTH-independent from ACTH-dependent causes | 1. Unaffected by changes in cortisol binding proteins | 1. Easy administration | |
2. Reflects 24 h total cortisol and not just a single time point | |||
Disadvantages | 1. Interpretation complicated by medications altering dexamethasone metabolism | 1. Difficult to collect | 1. Need different measurement time if shift worker, or has sleep disorder |
2. False positives in pregnancy/hormonal therapy | 2. Sensitivity and specificity affected by renal function and volume of urine | 2. Need to avoid licorice, tobacco | |
3. Unreliable in renal failure | |||
Criteria for positive result | < 1.8 µg/dL (higher specificity) < 5 µg/dL (higher sensitivity) | Upper limit of normal for institutional assay | > 145 ng/dL |
Pseudo-Cushing’s States
An important limitation of all tests for CS is the difficulty distinguishing true CS from conditions that lead to a temporary increase in cortisol (pseudo-Cushing states). Significant alcoholism, depression, panic attacks, obesity, stress, and intensive care unit (ICU) admission or critical illness can all transiently increase cortisol levels and lead to false-positive results. Additionally, the use of estrogens or oral contraceptives leads to increase in serum cortisol via higher levels of binding proteins (see section on UFC below). When attempting to diagnose CS, the presence of pseudo-Cushing states need to be considered and addressed when possible.
Low-Dose Dexamethasone Suppression Test
The rationale behind dexamethasone suppression testing is that the normal HPA responds to an increase in corticosteroids by suppressing ACTH secretion which leads to a drop in cortisol. In patients with ACTH-independent CS, the addition of dexamethasone should have minimal impact on serum cortisol because the feedback effect of excess cortisol has already suppressed ACTH secretion. Similarly, ectopic sources of ACTH secretion are generally not inhibited by receiving dexamethasone. Pituitary adenomas also tend not to be completely suppressed by low doses of dexamethasone, though there is often at least some decrease in serum cortisol after administration of dexamethasone. In practice, some patients with adrenal and even ectopic ACTH secretion (EAS) will have cortisol suppression in response to the LDDST. There are several methods for conducting the LDDST but one common approach involves oral administration of 1 mg dexamethasone between 2300 and 2400 h followed by plasma cortisol measurement between 0800 and 0900 h that morning. Overall sensitivity of LDDST may be as high as 95 % with a cutoff point of 1.8 µg/dL, though there is some disagreement over where this threshold should be set. Using a higher cutoff of < 5 µg/dL will decrease sensitivity but fewer false positives results in improved specificity [19–26].
The results of a LDDST can be affected by any of the pseudo-Cushing states mentioned above as well as by any process or medication that alters dexamethasone metabolism. These are discussed in more detail in a later section. Notably, women taking hormone supplementation may have up to a 50 % false-positive rate.
Twenty-Four-Hour UFC
Serum cortisol circulates in an inactive form bound to cortisol-binding protein. The active or free cortisol is a function of both total cortisol levels and concentration relative to binding proteins. Serum measurements are affected by medications and comorbidities that influence cortisol-binding globulin (CBG) levels. These include use of oral contraceptives by women as estrogen increases CBG production and this leads to elevated serum cortisol measurement. By contrast, UFC reflects the quantity of unbound cortisol that is filtered through the kidney and does not fluctuate with CBG concentration. Since UFC is collected over a 24-h period, it provides an overall assessment of free cortisol levels rather than relying on detecting elevated cortisol at a single time point. Sensitivity and specificity vary somewhat depending on the threshold set for an abnormal result but are typically > 85 and 90 %, respectively [27, 28]. There are several limitations to measuring UFC. First, since the assay depends on renal filtration, alterations in kidney function can significantly alter cortisol levels. At a creatinine clearance of < 60 cc/min, the rate of false negatives begins to increase substantially as less cortisol is filtered and collected in the specimen. At the opposite end of the spectrum, a substantial increase in fluid intake can lead to falsely elevated UFC as renal filtration increases. Additionally, performing the test can be something of a burden as patients need to provide a complete 24-h sample for the assay to be reliable.
Late-Night Salivary Cortisol
In otherwise healthy individuals, serum cortisol begins to rise prior to waking and peaks between 7 and 9 a.m. Cortisol then steadily declines throughout the day and reaches a nadir around midnight. This normal circadian rhythm is often interrupted in patients with CS so that midnight cortisol is substantially elevated. Plasma cortisol equilibrates with salivary levels and the concentration is not substantially affected by the amount of saliva produced. Equally important, increases in serum cortisol are rapidly reflected by increases in salivary cortisol. Since measurements of salivary levels do not require needle sticks and blood draws, this assay is a convenient way to measure cortisol in an outpatient setting. When a threshold value of > 145 ng/dL is set, sensitivity and specificity are comparable to the previous assays and range from 92 to 100 % and 93 to 100 %, respectively [27–30].
Salivary glands express 11β-hydroxysteroid dehydrogenase type 2 which converts cortisol to cortisone; it is possible that medications or other substances which block the enzyme may have falsely elevated salivary cortisol. Substances such as licorice and tobacco fall into this category and should be avoided during testing. Since the assay functions based on assumptions of a normal circadian rhythm, patients who work in night shifts or have altered sleep–wake cycles should have the timing of the assay adjusted appropriately or the measurement will miss the actual nadir in cortisol.
Additional Confirmation of Elevated Cortisol
If one of the above high sensitivity tests are used to demonstrate elevated cortisol, it is important to confirm the diagnosis with additional testing. Two elevated tests are required to confirm the diagnosis. If the diagnosis is equivocal based on initial testing, additional advanced testing can help better identify patients who truly have CS. One option is use of the 48-h, 2 mg/day LDDST with or without addition of CRH stimulation. This test involves administering 0.5 mg doses of dexamethasone every 6 h for 48 h and then measuring serum cortisol 6 h later. The addition of 1 µg/kg intravenous (IV) ovine CRH 2 h after the last dose of dexamethasone may improve both sensitivity and specificity. The high-dose dexamethasone suppression test (HDDST) is another reasonable alternative though some studies suggest that this test adds little information compared to the LDDST [31]. Alternatively, the midnight serum cortisol test can help confirm the diagnosis of CS [32]. This test requires inpatient admission and can be performed in patients while sleeping or while awake with different thresholds set for each approach.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


