Abstract
Hypopituitarism is defined as the deficiency of one or more of the anterior pituitary hormones: growth hormone, thyroid stimulating hormone, lutenizing hormone, follicle stimulating hormone, prolactin, and adrenocorticotrophic hormone. The deficiency of posterior pituitary hormones, specifically antidiuretic hormone, may also be present. Hypopituitarism is often a sequela of a previous neurological insult; however, many cases are considered idiopathic.
The development of the pituitary gland has been shown to be a balanced and orchestrated sequence of events dependent on the temporal and spatial appearance of multiple developmental factors that will eventually produce specific pituitary cell types. A disruption in this cascade that leads to the decreased expression of one or more of these factors results in idiopathic hypopituitarism. The identification of proteins responsible for the development of the anterior pituitary gland and their genetic alterations has provided novel insights into the mechanisms responsible for hypopituitarism. Murine models, as well as patients with hypopituitarism due to specific mutations in these developmental proteins, have assisted in the elucidation of mechanisms leading to decreased pituitary gene expression and hormone production. Furthermore, the development of more sophisticated and efficient methods for genome sequencing offers greater promise in delineating the pathogenic mechanisms in patients with idiopathic hypopituitarism.
Keywords
pituitary development, hypopituitarism, combined pituitary hormone deficiency
Introduction
Hypopituitarism is a condition that presents clinically with one or more deficiencies of hormones secreted from the adenohypophysis, neurohypophysis, or both. The annual incidence rate has been estimated to be 4.2 cases per 100,000 of the population, along with a prevalence of 45.5 per 100,000 in a 2001 population study from Spain. The etiology of hypopituitarism can be attributed to several causes including head injury, neurosurgical sequelae, infiltrative disorders, and cranial radiotherapy; however, for many patients, in particular for those with congenital or idiopathic hypopituitarism, the etiology for hormone deficiency cannot be identified. There appear to be limited data on the true prevalence of idiopathic hypopituitarism. The advancements in understanding hypothalamic-pituitary development using animal models have helped to identify a cascade of several developmental factors necessary for proper pituitary function. Mutations in these factors found in both animal models and affected patients have been identified and linked to the development of hypopituitarism.
Pituitary hormone deficiency may present in a wide variety of ways; for example, it may manifest acutely in neonates as an adrenal crisis or insidiously in children with a poor growth velocity. The clinical presentation will depend on the deficient hormone(s) and may be relatively nonspecific; symptoms may include increased lethargy, cold intolerance, poor weight gain, decreased appetite, or abdominal pain. An evaluation of pituitary function in the newborn period typically takes place in the setting of persistent hypoglycemia or electrolyte imbalance. Although the phenotype of idiopathic hypopituitarism is varied, infants found to have midline defects and male infants with micropenis should be evaluated for possible pituitary hormone deficiency. In children and adolescents, poor growth or delayed puberty is often an indicator for care providers to evaluate the pituitary for possible deficiencies. It is prudent to recognize that the evaluation of some patients may initially reveal a single pituitary hormone deficiency; however, follow-up evaluations are required, as additional pituitary hormone deficiencies may develop over time.
Genetic pathophysiology
The development of the pituitary occurs early during embryogenesis by the coordinated spatial and temporal expression of signaling molecules and transcription factors. Initially, the primordial Rathke’s pouch develops by the thickening and invagination of the oral ectoderm that comes into contact with the ventral diencephalon. Proliferation of the cells through expression of various signaling factors drives oral ectoderm closure and forms a detached rudimentary gland. The expression of Sonic hedgehog (Shh) from the oral ectoderm and downstream effectors, such as those in the Gli family, are important in orchestrating the proliferation and formation of the gland. In addition, the Wnt family of signaling molecules and bone morphogenetic protein 4 (BMP4), among others, also may have roles in guiding proliferation and specification of pituitary cell types.
The mature anterior pituitary gland ultimately contains five cell types regulated by trophic hormones produced by the hypothalamus, as well as positive and negative feedback from peripherally secreted hormones. The cell types and hormones they secrete include: somatotrophs, growth hormone (GH); thyrotrophs, thyroid-stimulating hormone (TSH), lactotrophs, prolactin (PRL), gonadotrophs, lutenizing hormone (LH), and follicle-stimulating hormone (FSH), and corticotrophs, adrenocorticotrophic hormone (ACTH). The development of these pituitary cell types is regulated by the expression of transcription factors including Hesx homeobox 1, LIM homeobox protein 3 (Lhx3), paired-like homeodomain 1 (Pitx1), and POU1F1, among others. These factors play significant roles in the coordinated temporal and spatial development of specific pituitary cell types ( Fig. 6.1 ). Since several of the pituitary cell types require common factors for their development, the early disruption in the cascade of events may ultimately affect several cell types. This may lead to loss of pituitary gene expression and secretion as well as abnormal structural development of the pituitary gland.
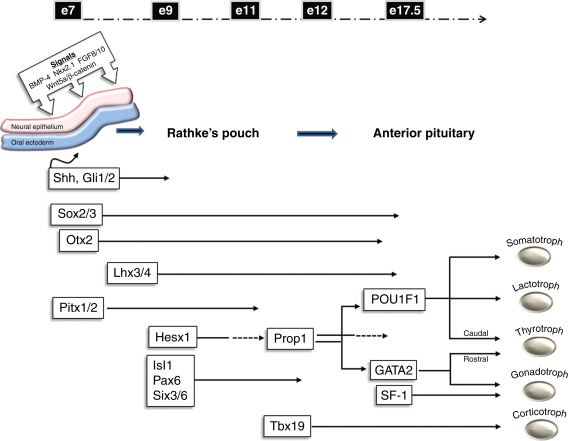
Although a large part of our knowledge regarding pituitary organogenesis is based on nonhuman studies, careful clinical investigation of patients with hypopituitarism has yielded important insights into pituitary development. For example, murine studies of the Ames dwarf mouse, which bears a mutation in the PROP1 gene, revealed a hypocellular anterior pituitary generally lacking somatotrophs, lactotrophs, and thyrotrophs. These findings led to studies in patients with a similar phenotype and the identification of human mutations in the PROP1 gene. Two other naturally occurring inbred murine models, the Jackson and Snell dwarf mice, illustrated the importance of POU1F1 (formerly Pit1) in normal pituitary development. Mutations in the POU1F1 gene were subsequently identified in patients and revealed the importance of heterozygous point mutations encoding proteins acting as dominant negative inhibitors of pituitary gene expression.
Over the last several decades, several research laboratories have shown that mutations in specific transcription factors can disrupt the balanced orchestration of pituitary development and ultimately the expression and function of the five pituitary cell types. This progress has resulted in the genetic characterization of hypopituitarism in many patients whose condition had been labeled as idiopathic or “congenital.” Mutations in several of these factors leads to multiple pituitary hormone deficiencies, whereas mutations in others may only lead to loss of a single hormone. Table 6.1 contains an outline of several genetic factors that have importance in anterior pituitary development. For each factor, the following are listed: a brief description of its function, the pituitary cell types affected by its disruption, a brief summary of the clinical features noted in patients with reported mutations, and the mode of inheritance. Several of these factors are associated with syndromes or are affected as a result of chromosomal abnormalities; thus, hypopituitarism may be one of the clues suggesting a genetic etiology. Finally, these factors are crucial to pituitary development, but may also be important for pituitary cell survival; therefore, the initial clinical phenotype in some patients may progress and additional pituitary hormone deficiencies can develop over time.
| Factor | Gene function | Affected cell types | Clinical phenotype | Mode of inheritance |
|---|---|---|---|---|
| One of more pituitary hormone deficiencies | ||||
| Hesx1 |
| Somatotrophs, thyrotrophs, gonadotrophs, corticotrophs, posterior pituitary may also be affected |
| AD, AR |
| Lhx3 (Lim3, P-LIM) |
| Somatrotrophs, lactotrophs, thyrotrophs, gonadotrophs, possibly corticotrophs |
| AR |
| Lhx4 |
| Somatrotrophs, lactotrophs, thyrotrophs, gonadotrophs, corticotrophs |
| AD |
| Six6 ( Optx2 ) |
| Somatotrophs, gonadotrophs |
| Unclear |
| Pitx2 (RIEG1) |
| Somatrotrophs, lactotrophs, thyrotrophs, corticotrophs, reduced expression of gonadotrophs |
| AD |
| Prop1 (prophet of Pit 1) |
| Somatrotrophs, lactotrophs, thyrotrophs, gonadotrophs, corticotrophs (delayed) |
| AR |
| POU1F1 (Pit1) |
| Somatotrophs, lactotrophs, thyrotrophs |
| AD, AR |
| Otx2 |
| Somatotrophs, thyrotrophs, corticotrophs, gonadotrophs |
| Unknown |
| SOX2 |
| Somatotrophs, gonadotrophs, and in animal models thyrotrophs |
| De novo |
| SOX3 |
| Somatrotrophs and/or possible deficiency in gonadotrophs and thyrotrophs |
| X-linked |
| Isolated hormone deficiency | ||||
| GLI2 |
| Somatotrophs (possible association with CPHD) |
| |
| GHRHr |
| Somatrotrophs |
| AR |
| GH1 |
| Somatotrophs |
| AR, AD, or X-linked |
| KAL-1 |
| Gonadotrophs |
| X-linked |
| FGFR-1 (KAL-2) |
| Gonadotrophs |
| AD |
| FGF8 |
| Gonadotrophs |
| |
| PROKR2 PROK2 |
| Gonadotrophs |
| AR |
| GnRHRI |
| Gonadotrophs |
| AR |
| TBX19 (TPIT) |
| Corticotrophs |
| AR |
| POMC |
| Corticotrophs |
| AR |
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


