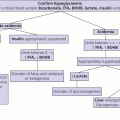
9 Regulation of salt balance is achieved primarily through activation of the renin–angiotensin–aldosterone system and the release of atrial natriuretic peptide. Renin is secreted by the juxtaglomerular cells of the kidney in response to sodium depletion or extracellular fluid volume restriction. Renin converts angiotensinogen to angiotensin I which, in turn, is converted by angiotensin‐converting enzyme to angiotensin II. Angiotensin II stimulates the production of aldosterone from the zona glomerulosa of the adrenal cortex. Adrenocorticotrophic hormone (ACTH) does not play a role in the physiological regulation of aldosterone secretion although serum aldosterone increases upon acute intravenous (IV) ACTH administration. Potassium ions also facilitate the secretion of aldosterone. By contrast, the secretion of both renin and aldosterone may be inhibited by atrial natriuretic peptide. Aldosterone binds to the mineralocorticoid receptor, resulting in increased reabsorption of sodium in the kidney, sweat and salivary glands. Sodium ions are exchanged for potassium and hydrogen ions in the distal tubule. Cortisol also has a strong binding affinity for the mineralocorticoid receptor but is prevented from doing so as a result of metabolism to inactive cortisone by 11‐β‐hydroxysteroid dehydrogenase‐2 in aldosterone‐sensitive tissues. Water balance is maintained by the interrelation between thirst, renal function, and the antidiuretic hormone arginine vasopressin (AVP). Vasopressin is synthesized in the supraoptic and paraventricular nuclei of the hypothalamus in a pre‐pro‐hormone form consisting of vasopressin, neurophysin II, and copeptin. These peptides are then cleaved during transport along the supraoptic–hypophyseal tract to be stored in the posterior pituitary. Vasopressin release is regulated by osmoreceptors in the hypothalamus which detect changes in plasma osmolality from 280 to 295 mOsm/kg as may occur with loss of extracellular water. High concentrations of vasopressin may also be secreted following baroreceptor‐detected reductions in blood volume or blood pressure of 5–10%. Baroreceptors are located in the carotid arch, aortic sinus, and left atrium and modulate vasopressinergic neuronal function via vagal and glossopharyngeal stimulation of the brainstem. Vasopressin binds to a V2 receptor in the renal collecting tubule which regulates the insertion of water channel proteins (aquaporin 2) into the cell membrane. These allow water to flow along an osmotic gradient from the tubular lumen into the cells lining the collecting duct. Other aquaporins (aquaporin 4) allow this water to pass to the renal interstitium and circulation. This regulatory mechanism maintains plasma osmolality between 282 and 295 mOsm/kg. When the plasma osmolality exceeds 295 mOsm/kg, vasopressin secretion cannot be increased further and fluid balance is maintained by increased thirst leading to increased fluid intake. The vasopressin effect is under negative feedback modulation by locally generated prostaglandins in the medullary collecting duct cells. Glucocorticoids are also required for free water excretion so that initiation of glucocorticoid replacement in hypopituitary patients may unmask diabetes insipidus (DI). Hyponatraemia may occur either as a result of salt and water depletion in which salt loss exceeds water loss or following fluid overload resulting in relatively more water than salt. The general mechanisms for the development of hyponatraemia are shown in Table 9.1. Table 9.1 Causes of hyponatraemia. Hyponatraemia associated with extracellular fluid loss is not always a direct consequence of the fluid loss per se, which is frequently hypotonic or isotonic by comparison with plasma, but may be caused by replacement of these fluid losses with hypotonic fluid (e.g. drinking water alone or use of hypotonic IV fluids). When a child presents with hyponatraemia for which the cause is not immediately apparent, the following points should be highlighted in the history: The following points should be highlighted in the clinical examination: Table 9.2 Clinical signs of volume depletion and overload. Hyponatraemia can be spurious either as a result of contamination of the blood sample taken from an IV cannula with hypotonic IV fluids or because of interference with the flame photometer assay by excess serum lipids or proteins. The key requirement in the assessment of a patient with hyponatraemia is to distinguish between causes associated with excess sodium loss and those associated with water retention, for example, in SIADH. The clinical distinction between these two states is summarized in Table 9.2. If the cause of the hypo‐osmolar state is not clear at presentation, urine osmolalities >100 mOsm/kg associated with urinary sodium concentrations >20 mmol/L suggest acute SIADH or renal, adrenal, or cerebral salt wasting. Urine osmolalities >100 mOsm/kg associated with urinary sodium concentrations <20 mmol/L suggest hypovolaemia or longer‐standing SIADH. Plasma renin is usually suppressed in SIADH but elevated in hypovolaemia. The causes of hyponatraemia are summarized in Table 9.1. Mineralocorticoid deficiency may be a consequence of idiopathic congenital adrenal hypoplasia or aplasia, biosynthetic defects of aldosterone synthesis (e.g. congenital adrenal hyperplasia) or acquired primary adrenal failure (e.g. Waterhouse–Friderichsen syndrome, autoimmune disease, or following surgical removal). While combined mineralocorticoid and glucocorticoid deficiency will cause hyponatraemia through salt loss, glucocorticoid deficiency (e.g. in hypopituitarism) will cause hyponatraemia due to impaired water excretion. Resistance to aldosterone may occur as a result of inactivation of the mineralocorticoid receptor or of the epithelial sodium channel. Abnormalities of mineralocorticoid physiology are discussed in more detail in Chapter 8. The various causes of SIADH are summarized in Table 9.3. Table 9.3 Causes of syndrome of inappropriate antidiuretic hormone secretion (SIADH). Where hyponatraemia is a consequence of sodium loss and in the context of clinical signs of significant hypovolaemia, IV colloid or 0.9% saline should be given until there is clinical evidence of circulatory improvement. Adrenal insufficiency should be treated with fludrocortisone and glucocorticoids (see Chapter 8). SIADH should be anticipated in individuals who have experienced significant head trauma or intracranial surgery and careful postoperative supervision of fluid balance is required. SIADH should be treated by fluid restriction which may range from only 40% to two‐thirds of normal intake. Where severe or symptomatic hyponatraemia or excessive thirst makes this approach impractical, then treatment to either increase water excretion or to raise the plasma sodium should be used. Water excretion will be enhanced by the tetracycline antibiotic demeclocycline which impairs the renal response to vasopressin and has been used in adults, giving 3–5 mg/kg eight‐hourly. An alternative is the relatively newly available V2 receptor antagonist tolvaptan, though trials of its efficacy in children are ongoing. Plasma sodium can be raised using hypertonic (3%) saline (0.1 ml/kg/min for two hours), aiming to increase plasma sodium concentration by about 10 mmol/L. In this context, it may be necessary to give furosemide with replacement of excreted urinary electrolytes to prevent hypervolaemia. This treatment should be reserved for those with significant neurological symptoms following the relatively acute onset of SIADH, as there is a risk of lethal pontine myelinolysis if serum sodium concentrations rise too rapidly (>10 mmol/L per day). Hypertension in childhood as a result of endocrine pathology is usually a consequence of either glucocorticoid or catecholamine excess, as shown in Table 9.4. Table 9.4 Causes of endocrine hypertension. Key points to highlight in the history and on clinical examination include the following: The following preliminary investigations should be considered if an endocrine cause of hypertension is suspected: If Cushing’s syndrome seems likely, additional investigations to confirm the diagnosis and treatment are described (see Chapter 8). If the urinary excretion of catecholamine metabolites is increased, then a blood sample should be taken for the measurement of free metanephrine. Two‐thirds of phaeochromocytomas are located in the adrenal medulla but they may also be found anywhere in the sympathetic chain, most commonly close to the renal hilum or aortic bifurcation. Abdominal imaging, preferably with computerized tomography (CT), magnetic resonance imaging (MRI), 123I‐metaiodobenzylguanidine (MIBG) scanning and, possibly, selective venous catecholamine sampling by catheterization may be necessary to locate the site(s). The various causes of endocrine hypertension are shown in Table 9.4. Hypertension in 11β‐hydroxylase‐ and 17α‐hydroxylase‐deficient congenital adrenal hyperplasia results from accumulation of the potent mineralocorticoid deoxycorticosterone, resulting in sodium and water retention with suppression of renin and aldosterone. 11β‐hydroxylase deficiency is also associated with excess androgen production and virilization, whereas 17α‐hydroxylase deficiency leads to female external genitalia in 46,XY subjects and lack of development of secondary sexual characteristics in both sexes. Primary aldosteronism is associated with hypernatraemia, increased plasma volume, hyporeninaemia, and hypokalaemia. Hypertension is common in childhood Cushing’s syndrome. The syndrome of apparent mineralocorticoid excess (AME) is characterized by low plasma renin and aldosterone concentrations and is associated with a deficiency of 11β‐hydroxysteroid dehydrogenase 2 which is responsible for metabolizing cortisol to cortisone to prevent high concentrations of cortisol from binding to the mineralocorticoid receptor. Liddle’s syndrome arises from an abnormality of renal tubular transport caused by an activating mutation of the amiloride‐sensitive sodium channel, resulting in increased sodium reabsorption and potassium loss with a biochemical and clinical picture similar to that of AME. Glucocorticoid‐suppressible hyperaldosteronism is a rare disorder in which primary aldosteronism is regulated by ACTH rather than renin–angiotensin because of the fusion of regulatory sequences of the 11β‐hydroxylase gene to coding sequences of the aldosterone synthase gene. In 11β‐hydroxylase‐ and 17α‐hydroxylase‐deficient congenital adrenal hyperplasia, hypertension responds to glucocorticoid therapy which suppresses ACTH secretion and thus deoxycorticosterone production. The treatment of Cushing’s syndrome is discussed in detail in Chapter 8. A phaeochromocytoma requires surgical removal in an experienced specialist centre with skilled anaesthetic support. Pre‐ and perioperative control of blood pressure must be achieved by the initial use of an adequate alpha blockade such as phenoxybenzamine. As this is achieved, supplemental salt intake is needed to expand the extracellular fluid volume. Beta blockers are also necessary to treat alpha‐blocker‐induced tachycardia. When a neuroblastoma causes catecholamine‐induced hypertension, similar medical management will be necessary in the pre‐operative period. The mechanisms of hypernatraemia are shown in Table 9.5 and include gastrointestinal fluid loss in which relatively more water is lost than salt, excessive salt intake (e.g. due to deliberate poisoning as in Munchausen‐by‐proxy), decreased water intake through impaired thirst, and excessive renal water losses, including diabetes insipidus. It is important to recognize that some patients with neurological disability may have a combination of impaired thirst and central diabetes insipidus. Rarely, congenital adipsia/hypodipsia may be seen in the context of a mid‐line defect with single central incisor. Unexplained episodic hypernatraemia should raise the possibility of factitious illness caused by the deliberate administration of salt by the child’s parent or carer. Table 9.5 Causes of hypernatraemia. Diabetes insipidus may occur either as a result of inadequate secretion of AVP (cranial or central diabetes insipidus) or when there is resistance to the antidiuretic effect of AVP (nephrogenic diabetes insipidus). Cranial diabetes insipidus may be congenital due to a gene defect or to a cerebral malformation (e.g. septo‐optic dysplasia (SOD), holoprosencephaly); or caused by acquired disease (e.g. craniopharyngioma, Langerhans cell histiocytosis, or surgery) of the hypothalamo–pituitary axis (Table 9.5). Autosomal dominant cranial diabetes insipidus may be caused by a mutation of the AVP–neurophysin II gene which leads to impaired processing of the AVP hormone precursor, causing progressive damage to the neurosecretory neurones of the hypothalamus and the development of increasingly severe symptoms of diabetes insipidus with advancing age. In the much less common autosomal recessive form, the symptoms occur earlier. Nephrogenic diabetes insipidus may occur as a consequence of mutations affecting the V2 receptor gene (X‐linked) or aquaporin 2 gene (autosomal recessive) or because of disorders of the kidney which impair the function of other components of the urinary concentrating mechanism (Table 9.5). The cardinal symptoms of diabetes insipidus are polyuria and polydipsia. Other causes of these symptoms must be considered – osmotic diuresis from glycosuria in diabetes mellitus and reduced nephron mass in chronic renal failure; excessive intake in habit drinking (psychogenic polydipsia); and impaired renal tubular function in hypercalcaemia and hypokalaemia. Additional clinical features of diabetes insipidus include constipation, fever, vomiting, loss of weight, failure to thrive, and dehydration. The following points should be highlighted in the history and clinical examination: Habitual excess drinking is common in toddlers and preschool children, and if often part of a wider management problem including a poor sleeping pattern. The child is otherwise healthy and the problem can usually be both diagnosed and cured by asking parents to stop flavoured fluids but allow the child unrestricted access to water. If symptoms persist, the child should be admitted for observation and the severity of the polyuria and polydipsia be confirmed by measurement of the 24‐hour fluid intake and urinary losses. A fasting blood sample should be taken for the measurement of plasma glucose and serum sodium, potassium, calcium, and creatinine concentrations. Urine should be tested for glycosuria and proteinuria. In a significantly symptomatic individual, simultaneous early morning blood and urine samples should be taken for the measurement of serum electrolytes and osmolality and urinary osmolality. Diabetes insipidus may be confirmed by the presence of a hyperosmolar state (i.e. serum osmolality >295 mOsm/kg) with inappropriately dilute urine (urine osmolarity around <750 mOsm/kg). The plasma or urine sample should then be sent for the measurement of AVP or plasma copeptin concentrations to confirm whether the cause is cranial or nephrogenic (Figure 9.1). In these circumstances, a water deprivation test would be dangerous and is contraindicated. Furthermore, a water deprivation test is not required when there is a clear history of polydipsia and polyuria in the context of underlying disease or treatment (e.g. craniopharyngioma, histiocytosis, and postoperative phase of craniopharyngioma) which is known to cause cranial diabetes insipidus. This test is time‐consuming for staff and unpleasant for the child and family. It can usually be avoided, being either unnecessary (young child with habit drinking), unsafe (e.g. postoperative craniopharyngioma) or both. It should only be carried out after consultation with an experienced physician and must be undertaken with particular care in young children. The following protocol can be used: If a diagnosis of cranial diabetes insipidus is made, an MRI of the hypothalamo–pituitary axis should be performed as there may be a pituitary tumour or stalk abnormality. The serum tumour markers β‐human chorionic gonadotrophin (β‐hCG) and α‐foetoprotein should also be measured. If the MRI demonstrates thickening of the pituitary stalk, repeat scans should be performed over the next several years to monitor the development of infiltrative disorders, such as Langerhans cell histiocytosis or a germinoma, especially if symptoms, such as headache or additional pituitary hormone deficiencies develop. Tests of wider anterior pituitary function may also be indicated. Diabetes insipidus may be masked by concurrent glucocorticoid insufficiency so that glucocorticoid replacement should be instituted before diagnostic tests for diabetes insipidus are performed. If during the water deprivation test, the plasma osmolality remains between 282 and 295 mOsm/kg and the urine osmolality increases to >750 mOsm/kg, the patient does not have diabetes insipidus and the possibility of primary polydipsia because of abnormal drinking habits should be considered. A diagnosis of cranial diabetes insipidus is suggested by the development of increased plasma osmolality >295 mOsm/kg in the presence of a urine osmolality <300 mOsm/kg which is then increased to >750 mOsm/kg following the administration of DDAVP. Failure of the urine to respond to DDAVP is indicative of nephrogenic diabetes insipidus. A partial urinary response (300–750 mOsm/kg) to water deprivation or DDAVP suggests partial cranial or nephrogenic diabetes insipidus. This should be treated with the long‐acting AVP analogue DDAVP and the following preparations are available: Widely varying dose regimens may be required, usually in two or three divided doses. The complete replacement dose of intranasal DDAVP is around 15 μg/m2 per day, and 10 μg of nasal DDAVP is roughly equivalent to 100 μg of oral DDAVP and to 60 μg of sublingual DDAVP. In those individuals taking DDAVP by the nasal route, an increase in the dosage of medication may be required during upper respiratory tract illnesses which may cause congestion of the nasal mucosa and impaired drug absorption. For this reason there has been a shift from nasal to oral DDAVP and, in recent years, a further move towards the sublingual preparation. Patients with cranial diabetes insipidus fall into three broad categories: postoperative craniopharyngioma patients who require very careful monitoring; cranial diabetes insipidus with intact thirst; and cranial diabetes insipidus with impaired thirst. This should be managed by treatment of any underlying metabolic cause. In the absence of this, treatment with indomethacin (0.5–1.0 mg/kg twice daily) and/or a thiazide diuretic (e.g. hydrochlorothiazide 0.5–1.0 mg/kg twice daily from birth to 12 years of age, or 12.5–25 mg twice daily in older children) together with a potassium‐sparing diuretic such as amiloride (5–10 mg/1.73 m2 twice daily) can be tried. Unfortunately, patients with nephrogenic diabetes insipidus often respond poorly to treatment and must be allowed adequate access to liberal amounts of water intake as required.
Salt and Water Balance
Physiology and pathophysiology
Control of salt balance
Control of water balance
Hyponatraemia
Aetiology
Mechanism
Examples
Salt loss
Renal disease
Renal tubular defects (e.g. Fanconi, Bartter syndromes)
Chronic renal failure
Interstitial nephritis
Renovascular hypertension
Diuretic treatment
Cisplatin toxicity
Aldosterone deficiency
Inherited enzyme disorders (e.g. 21‐hydroxylase deficiency), Addison’s disease
Aldosterone resistance
Pseudohypoaldosteronism
Cutaneous loss
Excess sweat sodium loss in cystic fibrosis, fluid loss in burns
Gastrointestinal
Vomiting and diarrhoea (e.g. in gastroenteritis)
Intestinal obstruction (e.g. intussusception)
Water excess
Renal disease
Acute nephritic syndrome, acute and chronic renal failure
Hypovolaemia or decreases in renal perfusion causing increased proximal renal tubular reabsorption
Cirrhosis, congestive heart failure, nephrotic syndrome
Excessive water intake
Iatrogenic – excessive intravenous fluid replacement
Primary polydipsia
ADH excess
Syndrome of inappropriate ADH secretion (e.g. in meningitis, pneumonia)
Over‐treatment with DDAVP
History and examination
Volume depletion
Volume overload
Investigations
Differential diagnosis
Diagnosis
Cause
Examples
Central nervous system disorders
Meningitis, encephalitis, trauma (including surgery), hypoxia, haemorrhage, ventriculo‐peritoneal shunt obstruction, Guillain–Barré syndrome
Respiratory disorders
Pneumonia, tuberculosis
Tumours
Thymoma, lymphoma, Ewing’s sarcoma
Drugs
Phenothiazines, tricyclic antidepressants, vincristine, narcotics DDAVP, prostaglandin synthetase inhibitors
Chlorpropamide, cyclophosphamide, carbamazepine
Treatment
Endocrine hypertension
Aetiology
Mechanism
Examples
Steroid‐mediated
Glucocorticoid excess
Iatrogenic (pharmacological doses, or over‐replacement in deficiency states)
Cushing’s syndrome
Apparent mineralocorticoid excess (AME)
Mineralocorticoid excess
11β‐hydroxylase deficiency, 17α‐hydroxylase deficiency
Liddle’s syndrome
Dexamethasone‐suppressible hyperaldosteronism
Catecholamine‐mediated
Phaeochromocytoma, paraganglioma, neuroblastoma
History and examination
Investigations
Diagnosis
Treatment
Hypernatraemia
Mechanism
Examples
Gastrointestinal fluid loss with relatively more water loss than salt loss
Gastroenteritis with hypertonic dehydration
Decreased water intake
Water deprivation
Impaired thirst
Excessive salt intake
Salt poisoning
Vasopressin deficiency (central diabetes insipidus)
Septo‐optic dysplasia, holoprosencephaly
Autosomal dominant vasopressin deficiency
Craniopharyngioma, germinoma, Langerhans cell histiocytosis, sarcoidosis
Autoimmune
Head injury, neurosurgery
Narcotic agonists
Vasopressin resistance (nephrogenic diabetes insipidus)
V2 receptor or aquaporin 2 gene defect
Nephrocalcinosis, nephronophthisis, polycystic kidney disease
Urethral valves
Hypercalcaemia, hypokalaemia
Lithium, demeclocycline, tolvaptan
Diabetes insipidus
Aetiology
History and examination
Investigations
Water deprivation test
Central nervous system testing and other investigations
Diagnosis
Treatment
Cranial diabetes insipidus
Nephrogenic diabetes insipidus
When to involve a specialist centre
Future developments
Controversial points
Potential pitfalls
Emergencies
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


