HEREDITARY DIFFUSE GASTRIC CANCER
Gastric cancer is the fourth most common cause of cancer worldwide and is the second leading cause of cancer mortality.
17 Although environmental agents, including
Helicobacter pylori and diet, are the primary risk factors for this disease, approximately 10% of gastric cancers are a result of familial clustering.
18,19 Histologically, gastric cancers may be classified as either intestinal or diffuse types. The intestinal type histopathology is linked to environmental factors and advanced age. The diffuse type occurs in younger patients and is associated with a familial predisposition. Because of a decrease in intestinal-type gastric cancers, the overall incidence of gastric cancer has declined significantly in the past 50 years. However, the incidence of diffuse gastric cancer (DGC), which is also called signet ring cell or linitis plastica, has remained stable and, by some reports, may be increasing.
Hereditary DGC (HDGC) is a genetic cancer susceptibility syndrome defined by one of the following: (1) two or more documented cases of DGC in first- or second-degree relatives, with at least one diagnosed before the age of 50; or (2) three or more cases of documented DGC in first- or second-degree relatives, independent of age of onset. The average age of onset of HDGC is 38, and the pattern of inheritance is autosomal dominant.
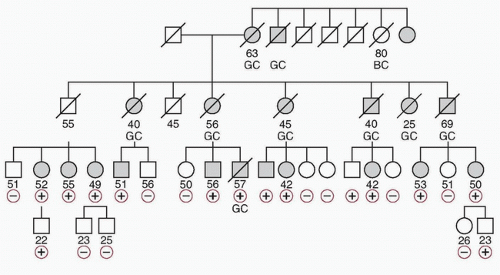
20 Figure 32.1 shows a pedigree with HDGC.
In 1998, inactivating germline mutations in the E-cadherin gene
CDH1 were identified in three Maori families, each with multiple cases of poorly differentiated DGC.
21 The
CDH1 mutations in these families were inherited in an autosomal dominant pattern, with incomplete but high penetrance. Onset of clinically apparent cancer was early, with the youngest affected individual dying of DGC at the age of 14.
21 Since then, germline mutations of
CDH1 have been identified in 30% to 50% of all patients with HDGC.
19,22 More than 50 mutations have been recognized across diverse ethnic backgrounds, including European, African American, Pakistani, Japanese, Korean, and others.
19 In addition to gastric cancers, germline
CDH1 mutations are associated with increased risk of lobular carcinoma of the breast, and this was the first manifestation of a
CDH1 mutation in one series.
23 CDH1 is, to date, the only gene implicated in HDGC. Penetrance of DGC in patients carrying a
CDH1 mutation is estimated at 70% to 80%, but may be higher. The need for a systematic study of specimens is supported by recent work by Gaya et al.
24 in which initial total gastrectomy specimens were reported as negative, but detailed sectioning and analysis showed invasive carcinoma.
CDH1 is localized on chromosome 16q22.1 and encodes the calcium-dependent cell adhesion glycoprotein E-cadherin. Functionally, E-cadherin impacts maintenance of normal tissue morphology and cellular differentiation. It is hypothesized that
CDH1 acts as a tumor suppressor gene in HDGC, with loss of function leading to loss of cell adhesion and subsequently to proliferation, invasion, and metastases.
Figure 32.2 shows the
CDH1 mutation for the pedigree depicted in
Figure 32.1.
The germline
CDH1 mutation is most frequently a truncating mutation. Germline missense mutations are causative in a few HDGC kindreds, but are more often clinically insignificant. In vitro assays for cellular invasion and aggregation may predict the functional impact of missense mutations to aid in this distinction.
22 Within the gastric mucosa, the “second hit” leading to complete loss of E-cadherin function results from
CDH1 promoter methylation, as has been described in sporadic gastric cancer.
25It remains unclear whether specific
CDH1 mutations are associated with distinctive phenotypic characteristics or rates of penetrance, although this may become apparent as more recurrent mutations are recognized. To date, most mutations identified have been novel and distributed throughout
CDH1. Recognition of recurrent mutations has usually resulted from independent events; however, there is evidence for the role of founder effects in certain kindreds.
22 At present, it is also unclear whether patients with HDGC without detectable
CDH1 mutations have mutation of a different gene or merely a
CDH1 mutation that has gone unrecognized.
New recommended screening criteria for CDH1 mutations are as follows:
Families with one or more cases of DGC
Individuals with DGC before the age of 40 years without a family history
Families or individuals with cases of DGC (one case below the age of 50 years) and lobular breast cancer
Cases where pathologists detect in situ signet ring cells or pagetoid spread of signet ring cells adjacent to diffuse type gastric cancer
18,26
As in other familial cancer syndromes, genetic counseling should take place prior to genetic testing so that the family understands the potential impact of the results. After obtaining informed consent, a team comprising a geneticist, gastroenterologist, surgeon, and oncologist should discuss the possible outcomes of testing and the management options associated with each. Genetic testing should first be performed on a family member with HDGC or on a tissue sample if no affected relative is living. In addition to direct sequencing, multiplex ligation-dependent probe amplification is recommended to test for large genomic rearrangements. If a
CDH1 mutation is identified, asymptomatic family members may proceed with genetic testing, preferably by the age of 20.
19 If no mutation is identified in the family member with DGC, the value of testing asymptomatic relatives is low.
Among individuals found to carry a germline
CDH1 mutation, clinical screening is problematic. Histologically, DGC is characterized by multiple infiltrates of malignant signet ring cells, which may underlie normal mucosa.
27 Because these malignant foci are small in size and widely distributed, they are difficult to identify via random endoscopic biopsy. Chromoendoscopy and positron emission tomography have reportedly been used, but the clinical utility of these tools in early detection remains unproven. Lack of a sensitive screening test for HDGC makes early diagnosis extremely challenging. By the time patients are symptomatic and present for treatment, many have diffuse involvement of the stomach or linitis plastica, and rates of mortality are high. Published case reports describe patients who have presented with extensive DGC despite recent normal endoscopy and negative biopsies.
28 The 5-year survival rate for individuals who develop clinically apparent DGC is only 10%, with the majority dying before age 40.
Because of high cancer penetrance, poor outcome, and inadequacy of clinical screening tools for HDGC, prophylactic total gastrectomy is recommended as a management option for asymptomatic carriers of
CDH1 mutations.
18 Although total gastrectomy is performed with prophylactic intent in these cases, most specimens have been found to contain foci of diffuse signet ring cell cancer.
19,28,29 Foci of DGC have been identified even in patients who have undergone extensive negative screening, including high-resolution computed tomography, positron emission tomography scan, chromoendoscopy-guided biopsies, and endoscopic ultrasonography.
19 However, HGDC in asymptomatic
CDH1 carriers is usually completely resected by prophylactic gastrectomy, as pathologic analyses of resected specimens have shown only T1N0 disease.
Because these signet ring cell cancers are multifocal and distributed throughout the entire stomach, especially in the cardia,
30 prophylactic gastrectomy should include the entire stomach, and the surgeon must transect the esophagus and not the proximal stomach. Furthermore, it should be performed by a surgeon experienced in the technical aspects of the procedure and familiar with HDGC. In asymptomatic patients, lymph node metastases have not been observed; therefore, D2 lymph node resection is not necessary. The optimal timing of prophylactic gastrectomy in individuals with
CDH1 mutations is unknown, but recent consensus recommendations indicate that age 20 is reasonable.
18Although it is a potentially lifesaving procedure, prophylactic gastrectomy for
CDH1 mutation carries significant risks that must be considered. Overall mortality for total gastrectomy is estimated to be as high as 2% to 4%, although it is estimated to be 1% when performed prophylactically. Patients must also be aware that there is a nearly 100% risk of long-term morbidity associated with this procedure, including diarrhea, dumping, weight loss, and difficulty eating.
19 A recent study of the effects of prophylactic gastrectomy for
CDH1 mutation demonstrated that physical and mental function were normal at 12 months, but specific digestive issues were recognized. Overall, 70% had diarrhea, 63% fatigue, 81% eating discomfort, 63% reflux, 45% eating restrictions, and 44% had altered body image, suggesting that this operation impacted negatively on quality of life.
31 Because of these complications and the fact that lymph node spread has not been observed, some recommend vaguspreserving gastrectomy done either open or laparoscopically. In addition, because the penetrance of
CDH1 mutations is incomplete, some patients who undergo prophylactic gastrectomy would never have gone on to develop clinically significant gastric cancer. Prophylactic gastrectomy has, in fact, been performed on several patients reported to show no evidence of gastric cancer on pathology.
29Some individuals with
CDH1 mutations choose not to pursue prophylactic gastrectomy. These individuals should undergo careful surveillance, including biannual chromoendoscopy with biopsies, beginning when they are at least 10 years younger than
the youngest family member with DGC was at time of diagnosis. It is recommended that any endoscopically visible lesion is targeted and that six random biopsies are taken from the following regions: antrum, transitional zone, body, fundus, and cardia. Careful white-light examination with targeted and random biopsies combined with detailed histopathology can identify early lesions and help to inform decision making with regard to gastrectomy.
32 Additionally, because women with
CDH1 mutations have a nearly 40% lifetime risk of developing lobular breast carcinoma, they should be carefully screened with annual mammography and breast MRI starting at age 35.
23 They should also do monthly self-examinations and have a breast examination by a physician every 6 months. The same surveillance recommendations are probably appropriate for HDGC families without identifiable
CDH1 mutations, although no current guidelines for this exist.
The emergence of gene-directed gastrectomy as a treatment strategy for patients with HDGC represents the culmination of a successful collaboration between molecular biologists, geneticists, oncologists, gastroenterologists, and surgeons. It is anticipated that the recognition of similar molecular markers in other familial cancer syndromes will transform the approach to the early diagnosis and treatment of a variety of tumors.
SURGICAL PROPHYLAXIS OF HEREDITARY OVARIAN AND ENDOMETRIAL CANCER
Hereditary Ovarian Cancer (BRCA1, BRCA2)
Inherited mutations in
BRCA1 and
BRCA2 strongly predispose women to breast cancer and to high-grade serous cancers of the ovary, fallopian tube, and peritoneum.
33,34 About two-thirds are due to
BRCA1 mutations and one-third
BRCA2 mutations, and these account for about 15% to 20% of high-grade serous cases. The lifetime risk of these gynecologic cancers increases from a baseline of 1.5% to about 15% to 25% in
BRCA2 carriers and 30% to 60% in
BRCA1 carriers.
33,34 BRCA1/2 mutations are rare in most populations (<1 in 500 individuals); one notable exception is the Ashkenazi Jewish population, in which the carrier frequency is 1 in 40.
35 BRCA1-associated cases peak in the 50s and
BRCA2-associated cancers in the 60s.
36 In addition to
BRCA1/2 mutations, germline mutations in a number of other genes in the homologous recombination DNA repair pathway confer high penetrance susceptibility to ovarian cancer (e.g.,
RAD51C,
RAD51D,
BRIP1,
PALB2).
37 This has led to the development of more comprehensive cancer genetic testing panels that are increasingly being used to identify women who are candidates for risk-reducing salpingooophorectomy (RRSO).
Genetic testing for inherited high-penetrance mutations in BRCA1/2 and other genes should be discussed with women who have a significant family history of early onset breast cancer and/or cancers of the ovary, fallopian tube, or peritoneum. Involvement of a genetic counselor prior to testing is helpful, as they have expertise in managing the inherent clinical and social issues. Most BRCA1/2 mutations involve base deletions or insertions in the coding sequence or splice sites that encode truncated protein products that are clearly dysfunctional. Less frequently, disease-causing mutations may occur that alter a single amino acid, though most of these missense variants represent innocent polymorphisms. The clinical significance of missense mutations can sometimes be elucidated by determining whether they segregate with cancer in other family members. In addition, genomic rearrangements may occur that inactivate BRCA1 or BRCA2, and identification of such alterations requires molecular testing beyond sequencing.
Penetrance of ovarian cancer is not 100% in those with clearly deleterious
BRCA1/2 mutations, but presently it is not possible to provide more precise personalized risk estimates to guide the use of RRSO. However, common variants have been discovered in other genes that appear to affect the risk of ovarian cancer in
BRCA1/2 carriers.
38 Based on the known ovarian cancer risk-modifying loci, it has been reported that the 5% of
BRCA1 carriers at lowest risk have a lifetime risk of ≤28% of developing ovarian cancer, whereas the 5% at highest risk have a ≥63% lifetime risk. In the future, when modifier loci are more completely catalogued, more precise estimates of cancer risk may be provided to individual patients who are considering RRSO.
As about 20% of women with high-grade serous ovarian cancers have
BRCA1/2 mutations, it has been suggested that all of these women undergo genetic testing regardless of family history.
39 Mutational analysis in women with these cancers may increasingly become standard practice as the cost of genetic testing declines. Testing may also be driven by the availability of poly(ADP-ribose) polymerase inhibitor therapy for women whose cancers have germline or sporadic mutations in genes such as
BRCA1/2 and others that are involved in homologous recombination DNA repair.
RRSO is strongly recommended in women who carry BRCA1/2 mutations because of the high mortality rate of ovarian cancer and the lack of effective screening and prevention approaches. Although screening with pelvic ultrasound and serum CA125 is generally recommended for BRCA1/2 carriers during their 20s and 30s, it is not proven to reduce ovarian cancer mortality because even early stage high-grade cancers have a very high mortality. Oral contraceptives reduce the risk of ovarian cancer in the general population and appear to have a similar effect in BRCA1/2 carriers, but this must be balanced against concerns regarding increased breast cancer risks.
The past practice of performing RRSO based solely on family history has been replaced by reliance on genetic testing. Clinical management of women with a strong family history in whom a deleterious germline mutation is not found, or those with variants of uncertain significance, should be resolved on a case-by-case basis. RRSO may be deemed appropriate in some cases, despite the absence of a clearly deleterious mutation. Fortunately, the risk of hereditary ovarian cancer does not rise dramatically until the mid-30s in women with
BRCA1 mutations and the 40s for women with
BRCA2 mutations.
36 As a result, most women are able to complete childbearing prior to undergoing RRSO. It is advisable for
BRCA1 carriers to undergo RRSO around age 35, as there is a 4% risk of ovarian cancer being discovered clinically or at the time of RRSO by age 40.
10 BRCA2 carriers may choose to delay surgery into their 40s due to their lower risk of ovarian cancer, but this could diminish the protection against breast cancer that is afforded by RRSO. If a mutation carrier, particularly a
BRCA1 carrier, chooses to pursue fertility into her 40s, then she should be counseled that she is at considerable risk of developing a life-threatening cancer that is largely preventable.
Several studies have provided evidence of the efficacy of RRSO. In one early study of
BRCA1/2 carriers, RRSO reduced the rate of breast and ovarian cancer by 75% over several years of follow-up.
40 A separate study in 2002 examined outcome in 551
BRCA1/2 carriers from various registries.
41 Among 259 women who had undergone RRSO, 6 (2.3%) were found to have stage I ovarian cancer at the time of the procedure and 2 (0.8%) subsequently developed serous peritoneal carcinoma. Among the controls, 58 (20%) women developed ovarian cancer after a mean follow-up of 8.8 years. With the exclusion of the six women whose cancers were diagnosed at surgery, RRSO reduced ovarian cancer risk by 96%. More recently, in 2014, an international registry study of over 5,783 subjects with median follow-up of 5.6 years found that RRSO reduced ovarian, tubal, and peritoneal cancer risk by 80%.
36 There was an estimated lifetime risk of primary peritoneal cancer after RRSO of about 4% for
BRCA1 carriers and 2% for
BRCA2 carriers.
36 The risk of death from all causes was reduced by 77%. A prospective cohort study noted that RRSO was associated with reduction in breast cancer-specific (hazard ratio [HR] = 0.44; 95% confidence interval [CI] = 0.26 to 0.76), ovarian cancer-specific (HR = 0.21; 95% CI = 0.06 to 0.80), and all-cause mortality (HR = 0.40; 95% CI = 0.26 to 0.61).
10Removal of the ovaries, as internal organs, usually has little effect on body image and self-esteem, and most BRCA1/2 mutation carriers elect to undergo RRSO. Insurance payers will almost always pay for RRSO in proven mutation carriers.
RRSO can be performed laparoscopically in most women, with discharge to home the same day. If a laparoscopic approach is problematic due to obesity or adhesions, the surgery can be performed through a small lower abdominal incision. Morbidity including bleeding, infection, and damage to the urinary or gastrointestinal tracts can occur, but the incidence of serious complications is very low. As the fallopian tubes and ovaries are small discrete organs, they are relatively easy to remove completely. Attention should be paid to transecting the ovarian artery and vein proximal to the ovary and tube so that remnants are not left behind. This involves opening the pelvic sidewall peritoneum, visualizing the ureter, and then isolating the ovarian blood supply. If there are adhesions between the adnexa and adjacent structures, careful dissection should be performed to ensure complete removal of the ovaries and fallopian tubes. If the uterus is not removed, care should be taken to remove the entire fallopian tube. A small portion of the tube inevitably will be left in the cornu of the uterus, but the risk of fallopian tube cancer developing in such remnants appears to be negligible.
Though there is not strong evidence that BRCA1/2 mutations increase uterine cancer risk, many women elect to have the uterus removed as part of the surgical procedure because they have completed their family or have other gynecologic indications. Although the addition of a hysterectomy may increase operative time, blood loss, surgical complications, and hospital stay, it usually can be performed laparoscopically and serious adverse outcomes are infrequent. Furthermore, the likelihood of future exposure to tamoxifen in the context of breast cancer prevention or treatment, which increases endometrial cancer risk two- to three-fold, also argues for concomitant hysterectomy. Women who receive hormone replacement therapy after surgery will require a progestin along with estrogen to protect against the development of endometrial cancer if the uterus is not removed.
In younger women, surgical menopause after RRSO is associated with vasomotor symptoms, vaginal atrophy, decreased libido, and an accelerated onset and incidence of osteoporosis and cardiovascular disease. In premenopausal women who do not have a personal history of breast cancer, estrogen replacement can be administered to ameliorate many of the deleterious effects of premature menopause. Systemic estrogen levels are lower in oopho-rectomized premenopausal women taking hormone replacement than if the ovaries had been left in place. The therapeutic benefit of oophorectomy in women with breast cancer has long been appreciated, and more recent studies support the contention that RRSO reduces the risk of breast cancer by about half in
BRCA1/2 carriers.
42 However, a meta-analysis showed that while RRSO was strongly protective against estrogen receptor-positive breast cancer (HR = 0.22), there was no protection against estrogen receptor-negative breast cancer.
42 Many carriers are identified after developing early onset breast cancer, and this group represents the most difficult in which to balance the potential risks and benefits of estrogen replacement therapy.
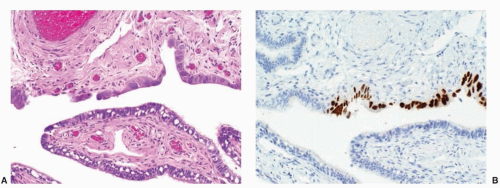
Early stage high-grade serous cancers and in situ lesions with
TP53 mutations have been identified in the fallopian tubes of some RRSO specimens (
Fig. 32.3). This has led to a paradigm shift in which it is now thought that most high-grade serous cancers found in the ovary, fallopian tube, and peritoneum are derived from cells that originate in the tubal fimbria.
43 The frequency of occult malignancies has varied between reports, but appears to be about 3%.
43 In view of this, the pelvis and peritoneal cavity should be examined carefully. Malignant cells also have been found in peritoneal cytologic specimens, and washings of the pelvis should be obtained when performing RRSO. The pathologist should be informed of the indication for surgery and serial sections of the fallopian tubes should be performed to look for the presence of early lesions. Patients found to have occult invasive high-grade serous cancers should be treated with chemotherapy after surgery. Those with in situ lesions appear to have a good outcome without chemotherapy.
44Cases of peritoneal serous carcinoma indistinguishable from ovarian cancer have been observed years after RRSO, but the origin of these cancers is unclear. Some may represent recurrences of occult ovarian or tubal cancers. In this regard, retrospective examination of the ovaries and fallopian tubes sometimes has revealed primary cancers that were not originally recognized. In contrast, some of these cancers likely arise directly from fallopian tube cells that have implanted in the peritoneum and subsequently become malignant. Patients who undergo RRSO should be made aware of their residual risk of peritoneal cancer, but there is no evidence that continued surveillance using CA125 and/or ultrasound is beneficial.