The recent approval of ruxolitinib (INCB018424) for myelofibrosis and the preclinical/clinical development of several additional janus kinase (JAK)-targeted agents have ushered in an era of novel therapies for advanced myeloproliferative neoplasms (MPN), which are associated with constitutive activation of the JAK–signal transducer and activation of transcription (STAT) signaling pathway. Collectively, these novel therapeutic approaches could rapidly broaden the spectrum of available therapies, with potential for improved clinical outcome for patients with advanced MPN. This review covers the recent developments in the testing of novel therapeutic agents other than JAK inhibitors that target signaling pathways in addition to JAK/STAT, or target the deregulated epigenetic mechanisms in MPN.
- •
With the recent approval by the US Food and Drug Administration of ruxolitinib, significant progress has been made in the treatment of myeloproliferative neoplasms.
- •
Drug toxicity and lack of effect on janus kinase 2 (JAK2) V617F allelic burden remain a problem with JAK inhibitors in the treatment of myeloproliferative neoplasms (MPN).
- •
Activation of collaborating pathways downstream of JAK2 V617F strongly supports the development and testing of novel agents for the treatment of patients with MPN.
- •
Treatment with chromatin-modifying agents (eg, histone deacetylase inhibitors and DNA methyltransferase 1 inhibitors) targets the deregulated epigenome and shows activity against MPN cells.
- •
Clinical experience with JAK inhibitors supports the rationale to develop safe and effective combinations, which incorporate JAK inhibitors with other agents active against MPN cells.
Introduction
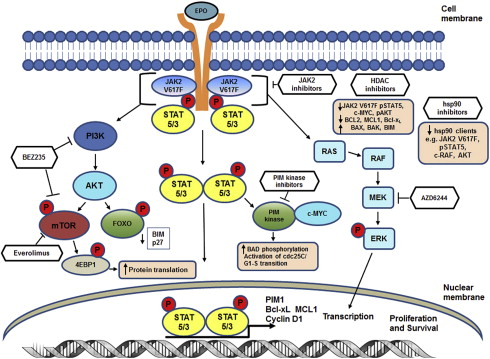
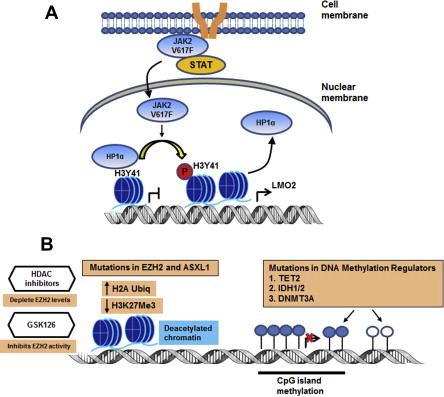
The identification of an acquired, somatic, gain-of function, point mutation at valine 617, V617F, in the janus kinase 2 (JAK2) gene was a major advance in the understanding of the clonal Philadelphia-chromosome negative myeloproliferative neoplasms (MPNs). The JAK2-V617F mutation is present in ∼90% of patients with polycythemia vera and in approximately 50% to 60% of patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF). JAK2 is a cytoplasmic, non-receptor, tyrosine kinase that via its association with cytokine receptors serves as a signaling mediator for hematopoietic cytokines such as erythropoietin (Epo) and thrombopoietin (Tpo) to regulate cell proliferation and growth ( Fig. 1 ). Binding of a ligand to cytokine receptor results in phosphorylation and activation of JAK2, which is the initiating step in the signaling cascade. On activation, JAK2 recruits and phosphorylates the signal transducer and activation of transcription (STAT) factors STAT5 and STAT3. This situation results in dimerization and translocation of STAT3 and STAT5 to the nucleus, where they activate transcription of genes involved in cell proliferation, survival, and resistance to apoptosis (eg, Bcl-xL, MCL1, PIM1, and Cyclin D1). JAK2 kinase activity is regulated by homology domains, in particular, the JH1 (catalytic kinase domain) and the JH2 (the pseudokinase domain), which serves an autoinhibitory function. The V617F point mutation within the JH2 domain disrupts its autoinhibitory function, resulting in constitutive activation of JAK2-mediated signaling pathways through the STAT5 and STAT3, phosphatidylinositol 3-kinase (PI3K) and extracellular signal-regulated kinase. Studies have shown that cytoplasmic STAT5a, through GAB2, is the link between JAK/STAT signaling and activation of the PI3K/AKT/mammalian target of rapamycin (mTOR) pathways. JAK2V617F-mediated alterations in transcription can also occur through a noncanonical pathway through which JAK2 enters the nucleus, phosphorylates histone H3 at tyrosine (Y) 41 and displaces HP1α from the chromatin. Phosphorylation of Y41 on histone H3 leads to increased expression of oncogenes such as LMO2 in MPN cells ( Fig. 2 A). Since the identification of JAK2V617F in MPN, additional mutations in JAK2 and other genes have been identified that also lead to constitutive activation of JAK/STAT signaling. One example is the identification of 4 novel somatic mutations in exon 12 of JAK2. These mutations are located in a region between the SH2 and JH2 domain and result in the gain-of function and activation of JAK2 similar to the V617F mutation. In JAK2V617F-negative MPN, mutations have also been described in the Tpo receptor c-MPL, particularly a point mutation at codon 515, which results in the substitution of tryptophan by leucine, lysine, asparagines, or alanine (W515L/K/N/A). Mutations in MPL are associated with spontaneous activation of MPL and downstream signaling through JAK/STAT. Similar to the action of constitutively activated JAK2V617F, expression of MPLW515L has been shown to transform hematopoietic stem cells (HSCs), resulting in growth factor independence, deregulated signaling through STAT, mitogen-activated protein kinase, and PI3K pathways as well as upregulation of anti-apoptotic proteins such as Bcl-xL. Previous studies have shown that expression of MPLW515L in murine models causes lethal MPN, with myelofibrosis (MF) resulting from JAK2 activation. Although less common (<10% of MPN cases), additional mutations in MPN have also been described in the intracellular signaling genes (eg, Casitas B-cell lymphoma [CBL]). CBL is an adapter protein that has also been shown to possess E3 ubiquitin ligase activity, and it serves to negatively regulate JAK2 signaling through ubiquitination and subsequent internalization of growth factor receptors. Because of these factors, CBL has been considered to be a tumor suppressor gene that acts to suppress JAK2 signaling. In MPN cells, mutations that inactivate or cause truncation of CBL result in enhanced downstream signaling through JAK/STAT and modulation of cell cycle, proliferation, and apoptosis-related proteins. Suppressor of cytokine signaling (SOCS) proteins are also negative regulators of JAK signaling, the expression of which is inactivated by mutation or through CpG island hypermethylation of the SOCS1 or SOCS3 promoter in MPN cells, leading to excessive cytokine signaling. Although mutations in the SOCS genes have been described in MPN, their occurrence is rare, and their role in the pathogenesis of MPN is controversial. In addition, LNK mutations have been described in MPN. LNK, (also called Src homology 2 B3), is a plasma membrane-bound adapter protein with several domains including an SH2 domain that can bind to MPL and JAK2 and negatively regulate JAK/STAT signaling in response to Epo receptor or MPL signaling. LNK mutations have been detected in 6% to 13% of chronic phase and blast phases of MPN and occur within a hot spot in the pleckstrin homology domain of exon 2. LNK mutations in JAK2V617F-negative MPN are associated with myeloid progenitor expansion and cytokine responsive pSTAT5 and pSTAT3 expression, which phenocopies activating mutations in JAK2.
Additional signaling pathways activated in MPN cells
In addition to the mutations activating the JAK/STAT pathway, other pro-growth and pro-survival signaling pathways downstream of the JAK/STAT pathway are also deregulated in MPN (see Fig. 1 ). These pathways includes the PI-3K/AKT/mTOR pathway, which cross talks and activates the RAS/RAF/MEK/ERK pathway, downregulating the pro-apoptotic protein BIM, in response to growth factor signaling in cancer cells. MEK is a key protein kinase in the RAS/RAF/MEK/ERK pathway, which signals for cancer cell proliferation and survival. MEK has been found to be frequently activated in cancer and regulates the biosynthesis of the inflammatory cytokines tumor necrosis factor, interleukin 6 (IL-6), and IL-1, which can act as growth and survival factors in cancer cells. Because the PI3K/AKT/mTOR pathway is commonly activated in many hematologic malignancies including myeloproliferative neoplasms, inhibition of this pathway could have broad anti-MPN activity. Constitutive, unregulated JAK/STAT activity can also result in activation of the proviral integration site (PIM) kinases. Expression of PIM1 is directly regulated through JAK/STAT signaling when the FERM domain of JAK2 is intact. The PIM kinases have been shown to promote the growth and survival of transformed myeloid progenitor cells.
Additional signaling pathways activated in MPN cells
In addition to the mutations activating the JAK/STAT pathway, other pro-growth and pro-survival signaling pathways downstream of the JAK/STAT pathway are also deregulated in MPN (see Fig. 1 ). These pathways includes the PI-3K/AKT/mTOR pathway, which cross talks and activates the RAS/RAF/MEK/ERK pathway, downregulating the pro-apoptotic protein BIM, in response to growth factor signaling in cancer cells. MEK is a key protein kinase in the RAS/RAF/MEK/ERK pathway, which signals for cancer cell proliferation and survival. MEK has been found to be frequently activated in cancer and regulates the biosynthesis of the inflammatory cytokines tumor necrosis factor, interleukin 6 (IL-6), and IL-1, which can act as growth and survival factors in cancer cells. Because the PI3K/AKT/mTOR pathway is commonly activated in many hematologic malignancies including myeloproliferative neoplasms, inhibition of this pathway could have broad anti-MPN activity. Constitutive, unregulated JAK/STAT activity can also result in activation of the proviral integration site (PIM) kinases. Expression of PIM1 is directly regulated through JAK/STAT signaling when the FERM domain of JAK2 is intact. The PIM kinases have been shown to promote the growth and survival of transformed myeloid progenitor cells.
Mutations in epigenetic modifiers in MPN
Similar to the other myeloid malignancies, mutations in genes regulating the epigenetic control of gene expression, the so-called epimutations, has also been noted in MPNs. These epimutations include mutations in ASXL1 (additional sex combs like 1), the polycomb group protein EZH2 and TET2 (ten-eleven-translocation 2) (see Fig. 2 B). The enhancer of zeste (EZH2) gene encodes a histone methyltransferase that along with suppressor of zeste 12 (SUZ12) and embryonic ectoderm development (EED) comprises the polycomb repressive complex (PRC) 2. The PRC2 complex functions to epigenetically silence gene expression through trimethylation of lysine (K) 27 on histone H3, a repressive histone mark. Mutations in EZH2 have been observed throughout the coding sequence but predominate in the C-terminal SET domain and negatively affect the histone methyltransferase activity of the PRC2 for lysine (K) 27 on histone H3. EZH2 mutations have been identified in 5.9% of patients with PMF, 1.2% of post-PV-MF, and 9.4% of post-ET-MF and were found to coexist in ∼40% of JAK2V617F-positive and ∼22% of ASXL1 mutation-positive patients with MF. Patients coexpressing these mutations cluster into the International Prognostic Scoring System (IPSS) high-risk category, have shorter leukemia-free survival and significantly shorter overall survival (OS) compared with EZH2 wild-type (WT) patients. Patients with PMF and mutated EZH2 showed higher leukocyte and blast counts and presented with larger spleens at diagnosis than those with WT EZH2. ASXL1 mutations have been identified in ∼55% of PMF and 22% of post-PV/ET-MF patients. Mutations in ASXL1 are most commonly located in exon 12 and result in a missense or a frameshift, resulting in a loss of function for ASXL1 and decreased ASXL1 expression. Loss of ASXL1 function leads to a genome-wide loss of trimethylation on lysine 27 of histone H3 and increased gene expression at loci with bivalent chromatin marks. ASXL1 mutations have also been observed to coexist with JAK2V617F in approximately 48% of PMF and post-PV/ET-MF patients. Similar to EZH2 mutations, patients with coexistence of ASXL1 mutations and JAK2V617F cluster into the IPSS high-risk category, experience significantly shorter LFS, and significantly shorter OS. In addition, ASXL1 mutant patients had a greater than 2-fold higher incidence of leukemia (34.9% vs 15.2%) than ASXL1 wild-type patients. Furthermore, LFS in double ASXL1/EZH2-mutated PMF patients was significantly shorter than in patients with single ASXL1 or EZH2 mutations (25 vs 138 vs 153 months). The TET2 gene is located on chromosome 4q24, a common break point involved in translocations in myeloid neoplasms. TET2 functions to convert 5-methylcytosine to 5-hydroxymethycytosine (5hmC) and has been found to be mutated in 7% to 16% of patients with JAK2V617F MPN. Mutations in TET2 compromise its catalytic activity, resulting in lower levels of 5-hmC in the genomic DNA as well as global hypomethylation of DNA. TET2 mutations have been observed in both JAK2V617F-positive and JAK2V617F-negative MPN; however, because of the diverse array of the identified mutations, they provide limited prognostic relevance. Similar to other myeloid malignancies, isocitrate dehydrogenase 1 (IDH1) and IDH2 mutations have been identified in MPNs. Unlike WT IDH1 and IDH2, which catalyze the conversion of isocitrate to α-ketoglutarate (α-KG), mutant IDH1 and IDH2 proteins possess neomorphic enzyme activity, resulting in the production of the oncometabolite, 2-hydroxyglutarate (2-HG). This enzymatic activity and the resulting 2-HG lead to impairment of TET2 catalytic function and global increases in DNA hypermethylation. Recently, 2-HG produced by IDH mutant cells was also shown to competitively inhibit α-KG-dependent dioxygenases, including the histone demethylation activity of a family of Jumonji-C domain histone demethylases. This situation led to marked increases in repressive histone methylation marks, suggesting that IDH mutations preferentially affect repressive histone methylation marks in addition to increasing DNA methylation in the transformed cells. Mutations in IDH1/2 are believed to be independent predictors of leukemic transformation because they are frequently identified in blast-phase MPN but not chronic-phase MPN. This finding suggests that IDH mutations may collaborate with JAK2V617F in transformation to leukemia. A recent study showed that IDH-mutated MPN patients showed significantly shorter LFS and OS than patients with mutated JAK2. DNMT3A belongs to a family of DNA methyltransferases that includes DNMT1 and DNMT3B, which catalyze the conversion of cytosine to 5-methylcytosine. DNMT3A mutations have been identified in MPN, although they are most commonly found in cases of MDS and acute myeloid leukemia (AML) at position R882, which lies in the methyltransferase domain. Previous studies have shown that loss of DNMT3A in HSCs resulted in altered DNA methylation patterns at different loci. DNMT3A mutations in myeloid malignancies lead to an impairment of its DNA methyltransferase activity, which may improve the response to treatment with DNA hypomethylating agents. Collectively, the documentation of epimutations in MPN creates a compelling rationale to test agents that target deregulated epigenetic mechanisms in MPN.
JAK2 kinase inhibitors in the clinic
Its crucial role in cytokine signaling in normal and malignant cells, and the observation that JAK2 mutations are present in most MPNs, prompted the development of several inhibitors of JAK (JAK- tyrosine kinase inhibitor [TKI]) (eg, ruxolitinib, SAR302503, CYT387, BMS911543, and CEP701) for the treatment of these neoplasms. INCB018424 (ruxolitinib) is a potent, small-molecule inhibitor of JAK1 and JAK2. A phase I-II study of ruxolitinib in MF showed that 15-mg twice-daily dosing was safe and effective in decreasing circulating inflammatory cytokines and improved constitutional symptoms. In 52% of treated patients, ruxolitinib caused rapid and sustained reduction in spleen size (≥50% reduction). Subsequently, 2 phase III studies of ruxolitinib were completed, which led to its approval by the US Food and Drug Administration (FDA) as a therapy for the treatment of MF. Another small molecule under clinical investigation for the treatment of MPN is SAR302503 (TG101348), a JAK2-specific inhibitor. Treatment with SAR302503 improved constitutional symptoms, and in 40% of patients with MPN caused a 50% or greater reduction in spleen size. SAR302503 treatment was also noted to significantly reduce the JAK2V617F allelic burden in patients with MPN. A more complete description of the clinical experience with these and other JAK-TKIs is given elsewhere in this issue by S. Verstovsek.
Although conferring clinical benefit in patients with advanced MPN, JAK inhibitor therapy has not reduced MF and normalized bone marrow histopathology, or significantly reduced the allelic burden of JAK2V617F and improved the survival of patients with MF. Suboptimal responses to JAK2 TKI may be caused by many factors, including complexity of clonal structure of the MPN, cell intrinsic mechanisms that confer JAK2 TKI resistance, additional mutations in the JAK2 domain that confer JAK2 TKI resistance, and the inability of JAK2 TKIs to target the stem cells in MPN. In addition, mutations in epigenetic regulators may contribute to the aggressiveness and treatment refractoriness of advanced MPNs. These factors provide a compelling rationale to develop and test novel agents that are active on targets or pathways that collaborate with JAK/STAT-mediated progrowth and prosurvival signaling, or target the deregulated epigenome in advanced MPN. Clinical experience with JAK inhibitors thus far also supports the rationale to develop safe and more effective combinations of JAK-TKI with other active agents against MPN cells.
Additional novel agents for MPN therapy
Recently, numerous novel agents targeting diverse signaling targets in MPN cells have been developed and tested preclinically in the in vitro and in vivo models of MPN. These agents, including mTOR (TORC1 and TORC2) inhibitor, dual PI3K/mTOR inhibitor, AKT inhibitor, MEK inhibitor, PIM kinase inhibitor, histone deacetylase (HDAC) inhibitor, DNMT1 inhibitor, and hsp90 inhibitors, have been developed and evaluated as single agents and in some cases in combination with JAK2 inhibitors, with the goal of increasing the therapeutic options for patients with MPN ( Tables 1 and 2 ).
| Drug | Type of Inhibitor | Disease | Preclinical or Clinical | Reference |
|---|---|---|---|---|
| Everolimus | mTOR | JAK2 V617F and MPL MPN | Preclinical and clinical | |
| BEZ235 | PI3K/mTOR | JAK2 V617F MPN | Preclinical | |
| MK-2206 | Allosteric AKT | JAK2 V617F MPN | Preclinical | |
| SGI-1776 | PIM kinase | JAK2 V617F MPN | Preclinical | |
| AZD1208 | PIM1, PIM2, PIM3 kinase | Not tested in MPN | Preclinical | |
| Selumetinib (AZD6244) | MEK1 kinase | JAK2 V617F MPN | Preclinical | |
| Panobinostat | Pan-HDAC | JAK2 V617F MPN, PMF | Preclinical and clinical (phase I and phase II) | |
| Givinostat (ITF2357) | Class I/II HDAC | PV, ET, PMF | Preclinical and clinical (phase II) | |
| AUY922 | hsp90 | JAK2 V617F MPN | Preclinical | |
| PU-H71 | hsp90 | JAK2 V617F and MPL MPN | Preclinical | |
| PF-04449913 | Hedgehog | PMF | Clinical | |
| BC2059 | β-catenin antagonist | JAK2V617F MPN | Preclinical |
| Combination Therapy | Type of Inhibitor | Disease | Preclinical or Clinical | Reference |
|---|---|---|---|---|
| BEZ235 + TG101209 | PI3K/mTOR and JAK2-TKI | JAK2 V617F MPN | Preclinical | |
| Selumetinib + TG101209 | MEK kinase and JAK2-TKI | JAK2 V617F MPN | Preclinical | |
| SGI1776 + TG101209 | PIM kinase and JAK2-TKI | JAK2 V617F MPN | Preclinical | |
| Panobinostat + TG101209 | Pan-HDAC and JAK2-TKI | JAK2 V617F MPN | Preclinical | |
| Panobinostat + ruxolitinib | Pan-HDAC and JAK1/2-TKI | JAK2 V617F MPN | Preclinical (murine models) | |
| Givinostat + hydroxyurea | Class I/II HDAC and ribonucleotide reductase | PV | Clinical (phase II) | |
| Decitabine + vorinostat | DNMT1 and pan-HDAC | JAK2 V617F MPN | Preclinical (murine models) | |
| AUY922 + TG101209 | hsp90 and JAK-TKI | JAK2 V617F MPN | Preclinical | |
| BC2059 + TG101209 | β-catenin antagonist and JAK2-TKI | JAK2 V617F MPN | Preclinical |
mTOR and Dual PI3K/mTOR Inhibitors
In addition to constitutive and enhanced JAK/STAT signaling, MPN cells also show activation of the mTOR pathway downstream of PI3K/AKT. The mTOR pathway serves a critical role in multiple cellular processes including cell growth, proliferation, and survival. These factors make targeting the mTOR pathway an attractive therapeutic strategy for MPN. Previous studies have shown that treatment with the mTOR inhibitor, everolimus (RAD001), inhibited cell proliferation and induced apoptosis of cultured MPN cells, as well as preventing the growth of hematopoietic progenitor cells (HPCs) derived from patients with PV and PMF. The clinical activity of everolimus against MPN has also been documented. A phase I/II study with everolimus was performed in patients with JAK2V617F or MPL mutations as well as patients with WT JAK2 with intermediate-risk/high-risk scores. Patients were treated with 5 to 10 mg of everolimus per day. No dose-limiting toxicity was observed up to a dose of 10 mg/d. Although everolimus had no effect on the JAK2V617F allelic burden, 69% of patients experienced a complete resolution of systemic symptoms and 80% had reduction in pruritus. According to International Working Group for MPN Research and Treatment criteria for disease response, everolimus treatment produced a 23% response rate in the 39 patients enrolled on the study. In addition, overall clinical improvement was noted in 6 of 39 patients (∼20%) and 1 patient obtained a partial remission. These findings support the rationale for determining the clinical efficacy and safety of combined treatment with everolimus and ruxolitinib in patients with MF. There has also been considerable interest in targeting both the PI3K and mTOR pathways with dual PI3K/mTOR pathway inhibitors, several of which are in clinical development. Because MPN cells have aberrant activation of the PI3K and mTOR pathways downstream of JAK2, and considerable crosstalk occurs between the pathways, this makes dual targeting of PI3K and mTOR an attractive and rational strategy for the treatment of MPNs. Preclinical studies have shown that treatment of cultured and primary MF-MPN cells with the dual PI3K/mTOR inhibitor, BEZ235, induced cell cycle growth arrest and inhibited PI3K and mTOR signaling, shown by depletion of p-AKT, p-p70S6K, and p-4EBP1, as well as exerting lethal anti-MPN activity. This study also showed that the combination of BEZ235 and the JAK2-TKI TG101209 exerted synergistically lethal activity against cultured and primary CD34+ MF-MPN cells and relatively spared normal CD34+ bone marrow progenitor cells. These studies support the rationale to test the in vivo activity of BEZ235 alone or in combination with ruxolitinib against MPN.
AKT Inhibitors
Previous studies have shown that the PI3K/AKT pathway is activated and supports cell growth, proliferation, and survival of MPN cells. For this reason, targeting the activity of AKT could exert lethal effects against MPN cells. Preclinical studies have shown that treatment with an allosteric AKT inhibitor, MK-2206, attenuates pro-growth and pro-survival activity of AKT and induced proliferation arrest and apoptosis in cultured MPN cells. Treatment with MK-2206 also inhibited the in vitro colony growth of primary CD34+ MF-MPN cells. MK-2206 also showed in vivo activity against murine transplant models of MPN, manifesting as reduced peripheral blood (PB) leukocytosis and extramedullary hematopoiesis. These findings support the rationale to determine the in vitro and in vivo activity of JAK-TKI with AKT inhibitor against MPN cells.
MEK Inhibitors
Several studies have documented the increased activity of the RAS/RAF/MEK/ERK pathway in promoting growth and survival of MPN cells. Therefore, targeting MEK kinase to interrupt this signaling is likely to be a promising therapeutic strategy in MPN. Recent in vitro studies have shown that treatment with the MEK inhibitor selumetinib (AZD6244) in cultured and primary MPN cells depleted MEK1 kinase activity, as shown by reduced pERK1/2 expression. Treatment with selumetinib also induced apoptosis of cultured and primary CD34+ MPN cells. In addition, cotreatment with selumetinib and JAK2-TKI exerted synergistic apoptotic effects against cultured MPN cells. Recently, a phase II study with selumetinib was conducted in patients with AML. The study showed that daily administration of selumetinib was tolerated and exerted modest anti-leukemic activity in patients with AML. The safety profile and activity in AML support the rationale for testing selumetinib and other safe MEK inhibitors alone or in combination with JAK inhibitors in patients with MPN.
PIM Kinase Inhibitors
The oncoprotein PIM1 belongs to a family of evolutionarily conserved cytoplasmic serine/threonine kinases (PIM1, PIM2, PIM3) that have been implicated in hematologic maligancies. PIM kinases have been shown to be constitutively active, downstream effectors of JAK/STAT signaling that promote cell survival and proliferation and inhibit apoptosis by phosphorylating and inactivating the pro-apoptotic proteins BAD and ASK1 and through interaction with c-MYC. By phosphorylating PRAS40, an inhibitor of TORC1, PIM kinase activity dissociates PRAS40 from TORC1, resulting in activation of TORC1. PIM kinases are frequently overexpressed in hematologic malignancies including MPN, and because they do not require post-translational modifications, their activity is largely regulated at the transcriptional or post-transcriptional level. Preclinical studies have shown that the PIM kinase inhibitor SGI-1776 inhibited the activity of PIM kinase in cultured MPN cells, as shown by depletion of p-BAD (Ser112) expression, attenuation of PRAS40 phosphorylation, decreased activity of TORC1, and attenuation of p-4EBP1 levels. SGI-1776-mediated inhibition of PIM kinases also reduced c-MYC levels in MPN cells. Treatment with SGI-1776, as a single agent, induced apoptosis of cultured and primary MF-MPN cells, which was associated with induction of p27 and BIM levels. In addition, cotreatment with SGI-1776 was shown to enhance the anti-MPN activity of JAK2-TKI (TG101209) against cultured and primary MF-MPN cells. Another PIM kinase inhibitor, AZD1208, has shown in vitro and in vivo efficacy against other hematologic malignancies such as AML but has not yet been tested against MPN cells. Given that AZD1208 is a pan-PIM inhibitor, and because of its proven activity in acute leukemia cells, it is likely that such an agent would show similar activity against MPN. Collectively, these observations also support the testing of PIM kinase inhibitor alone or in combination with JAK-TKI in the treatment of MPN.
HDAC Inhibitors
MPN cells have been shown to have increased HDAC levels and enhanced HDAC activity, especially of class I HDACs (1, 2, and 8). Previous studies have shown that increased HDAC levels and activity strongly correlated with the degree of splenomegaly in MPN, particularly MF. The 18 known HDACs are grouped into 5 phylogenetic classes according to their sequence homology and the cofactor required for their catalytic activity. Class I (HDAC1-3 and HDAC1-8), class IIa (HDAC4, 5, 7, and 9), class IIb (HDAC6 and 10), and class IV (HDAC11) require a divalent metal ion (eg, zinc) for their deacetylase activity, whereas class III (also called sirtuins or SIRT1-7) are structurally and biochemically different from the other classes and are dependent on nicotine adenine dinucleotide (NAD+) for their catalytic activity. HDACs are known to play significant roles in the epigenetic regulation of gene expression not only through lysine deacetylation of histone proteins but also through regulating the acetylation status of nonhistone proteins such as transcription factors and the expression of microRNAs. HDAC inhibitors are novel, structurally diverse agents that can be divided into several structural classes, including hydroxamates, cyclic peptides, aliphatic acids, and benzamides. Most of the HDAC inhibitors that have been developed are pan-HDAC inhibitors that inhibit class I, class II, and class IV, although class I and class I/II-specific inhibitors such as romidepsin and givinostat have also been developed. By altering gene expression through chromatin-dependent and chromatin-independent mechanisms, regulating cell cycle progression, acetylation of proteins, inhibition of proliferation, and induction of cell death, HDAC inhibitors represent a promising class of agents for the treatment of hematologic malignancies including MPNs. Panobinostat (LBH589) is a potent hydroxamate-based pan-HDAC inhibitor with nanomolar inhibitor activity against class I, II, and IV HDACs, which has been shown to induce the acetylation of both histone and nonhistone proteins in transformed cells, deplete antiapoptotic proteins (eg, Bcl-x L , MCL1, and BCL-2) and induce pro-apoptotic proteins (eg, BAX, BAK, and BIM). Recent studies have determined the in vitro activity of panobinostat against MPN cells. Treatment with panobinostat depleted the expression levels of mutant JAK2V617F and inhibited JAK/STAT intracellular signaling in cultured and primary MF-MPN cells, induced cell cycle growth arrest, and upregulated prodeath proteins (eg, BIM). Panobinostat depleted JAK2V617F expression by disrupting its chaperone association with heat shock protein (hsp) 90, resulting in proteasomal degradation of JAKV617F protein. In addition, panobinostat was shown to deplete the expression levels of epigenetic regulators, including polycomb group proteins, EZH2, and SUZ12, and DNA methyltransferases in other myeloid malignancies (eg, AML), resulting in disassembly of the PRC2 complex and downregulation of trimethylated lysine 27 on histone H3. Panobinostat treatment was also shown to deplete the expression of the DNA methyltransferase, DNMT1, and disrupt its chaperone association with hsp90 and with EZH2 in myeloid malignancies. The clinical activity of panobinostat has also been tested against hematologic malignancies, including MPN. A recent phase I clinical trial was conducted with panobinostat in patients with hematologic malignancies, of which 13 had MF. Patients treated with panobinostat experienced clinical improvement, including reductions of spleen size (57%–86%). Improvement in disease-related symptoms were observed in 4 patients with MF at doses of 30 (n = 1) and 60 mg (n = 3) of administered orally 3 times a week. In a separate phase I study, prolonged, low-dose panobinostat (25 mg 3 times a week) ameliorated constitutional symptoms, improved clinical features, and caused significant reductions of marrow fibrosis. Preclinical studies have also shown that cotreatment with panobinostat further enhanced JAK2-TKI-mediated inhibition of JAK/STAT signaling in MPN and exerted synergistic anti-MPN activity against cultured and primary MF-MPN cells. The combination of panobinostat and ruxolitinib has also been studied in vivo and showed improved efficacy over either agent alone in mouse models of JAK2-V617F-driven disease, without notable toxicity. Based on these promising preclinical data, a phase I clinical trial has been initiated to test the combination of panobinostat with ruxolitinib in patients with MPN (NCT01433445). Preclinical studies have also shown the activity of other classes of HDAC inhibitors against MPN cells. Givinostat (ITF2357) is a synthetic class I/II HDAC inhibitor that has shown significant in vitro and in vivo activity against MPN cells, and that has exerted little toxicity against normal bone marrow progenitor cells, hepatocytes, and mesenchymal cells. Givinostat activity was greatest in MPN cells with expression of JAK2V617F compared with JAK2 WT-expressing cells. Based on the promising in vitro and in vivo preclinical data, a phase II study was conducted to evaluate the safety and efficacy of givinostat in 29 patients with PV, ET, or PMF. Givinostat was well tolerated and no grade IV toxicities were observed. Givinostat treatment alleviated pruritus in most patients, and 75% of patients with PV/ET and 38% of patients with MF had a reduction in spleen size. Of the 29 patients on the study, there was 1 complete response, 6 partial responses in patients with PV/ET, and 3 major responses in patients with MF. A phase II study was also conducted to test the efficacy of the combination of givinostat and the ribonucleotide reductase inhibitor, hydroxyurea (HU) in patients with PV. The combination of givinostat and HU was well tolerated and effective, with 50% of patients achieving a partial or complete response according to European LeukemiaNet criteria. In addition, the preclinical use of novel combinations using HDAC inhibitors and chromatin-modifying agents such as the DNA methyltransferase inhibitor decitabine (5-aza-2′-deoxycytidine) has been reported. In this epitargeted strategy for treating MF-MPN, the sequential treatment of decitabine followed by HDAC inhibitor (Zolinza [vorinostat]) or trichostatin A resulted in greater lethality of JAK2V617F+ PMF cells and spared normal CD34+ cells. These studies also identified that the combination of DNMT1 inhibitor and HDAC inhibitor led to significant reductions in the proportions of JAK2V617F HPCs as well as reductions in HPCs with chromosomal abnormalities. This combination also upregulated expression of the chemokine receptor CXCR4 on CD34+ cells, which restored their ability to respond to CXCL12 stimulation and home to the bone marrow rather than remain aberrantly mobilized. This epitargeted treatment strategy also reduced the short-term and long-term SCID repopulating cells and JAK2V617F+ HSCs in murine transplant models. These preclinical data create the rationale for epigenetic targeted therapy in the treatment of MPN. A phase II study was conducted with 5-azacytidine alone in patients with relapsed or refractory MF. The overall response rate on this study was 37%, and 2 patients obtained a partial or complete response. In addition, in the responders, a 61% mean reduction in circulating CD34+ cells was observed. Collectively, these preclinical and clinical data represent a novel therapeutic strategy for targeting MPN progenitor (HSCs) or potentially MPN stem cells. Recently, a novel inhibitor of EZH2 methyltransferase activity (GSK-126) was reported to inhibit the methyltransferase activity of WT EZH2 and EZH2 with activating mutations at Tyr641. Treatment with this inhibitor resulted in global reduction of H3K273Me and increased expression of EZH2 target genes as well as dose-dependent inhibition of tumor growth in mouse models of lymphoma. Although the spectrum of mutations in MPN differ from those in lymphomas, studies to determine the activity of GSK-126 alone or in combination with other agents such as HDAC inhibitors have yet to be conducted against MPN cells.
Hsp90 Inhibitors
Hsp90 is a highly conserved, homodimeric, ATP-dependent, abundantly expressed protein, which forms the core of a super chaperone complex. Hsp90 promotes the stabilization and folding of client proteins into their active conformation. Each monomer of hsp90 has a highly conserved amino terminal adenosine triphosphate-binding domain, a middle client protein-binding domain, and a carboxy terminal dimerization domain. Previous studies have shown that many oncoprotein kinases, including JAK2, FLT-3, and BCR-ABL, are client proteins of hsp90. Furthermore, previous studies have suggested that the mutant oncoprotein kinases are more dependent on the chaperone function of hsp90 than their unmutated counterparts. Many of the proteins that are deregulated and confer a pro-growth and pro-survival advantage to MPN cells (c-RAF, AKT, pSTAT5, and PIM) are also client proteins of hsp90. Several hsp90 inhibitors, tanespimycin (17-AAG), alvespimycin (17-DMAG), AUY922, and ganetespib (STA9090), have been developed and are in various stages of clinical development in the treatment of hematologic and epithelial malignancies. Preclinical studies have shown that inhibition of hsp90 in MPN cells also inhibits JAK2 expression and downstream signaling. A recent study determined the in vitro and in vivo effects of treatment with PU-H71, a novel, nonquinone-based hsp90 inhibitor against JAK2V617F-expressing and MPLW515L-expressing cells. Treatment with PU-H71 induced cell growth arrest and apoptosis of cultured and primary MPN cells. This finding was associated with inhibition of hsp90 chaperone function and loss of binding of JAK2 to hsp90. PU-H71 also depleted JAK2 expression levels and potently inhibited JAK2 downstream signaling in cultured and primary MPN cells. In vivo treatment of JAK2V617F or MPLW515L expressing mice with PU-H71 resulted in normalization of blood counts, decreased extramedullary hematopoiesis, and improved survival compared with vehicle control mice. Similar effects on JAK2V617F expression levels, downstream signaling and anti-MPN activity were also observed with another hsp90 inhibitor, ganetespib. Preclinical studies with AUY922, a third-generation hsp90 inhibitor, showed that hsp90 inhibition attenuated JAK2 V617F expression and inhibited JAK/STAT signaling in cultured and primary MPN cells. Treatment with AUY922 caused dose-dependent apoptosis of cultured and primary MPN cells. Furthermore, the combination of AUY922 and the JAK2-TKI, TG101209, increased JAK2 inhibitor-mediated depletion of JAK/STAT signaling and exerted superior anti-MPN cytotoxic activity against cultured and primary MF-MPN cells. In addition, this study also showed that the combination of AUY922 and TG101209 could overcome JAK-TKI resistance in cultured human MPN cells isolated under the selection pressure of a JAK inhibitor. AUY922 was also shown to be effective against JAK-TKI-resistant Ba/F3 cells expressing JAK2V617F in cis with other kinase domain mutations (eg, R683G, G935R, Y931C, and E864K) isolated by random mutagenesis in bacteria. Collectively, these studies support the rationale to test hsp90 inhibitors alone, or in combination with JAK-TKI for the treatment of MPN. Testing of hsp90 inhibitors is also merited in patients who develop resistance or intolerance to JAK-TKIs.
Hedgehog Inhibitor
Hedgehog (HH) signaling pathway is involved in the self-renewal of stem cells and hematopoiesis. HH signaling is activated by the HH ligand Sonic, Indian, or Desert, which negatively regulate the activity of the 12-span membrane receptor protein Patched (PTCH). In turn, PTC inhibits the 7-span transmembrane protein Smoothened (SMO). HH ligands bind to PTC, leading to its internalization and degradation, thereby releasing SMO to promote the dissociation of Suppressor of Fused (SUFU)-GLI complex. This situation results in nuclear localization and activity of GLI transcription factors (GLI1 and GLI2) and degradation of the repressor form GLI3. GLI1 and GLI2 induce the expression of the HH pathway target genes, including Cyclin D, Cyclin B, PTCH1, GLI1, GLI2, and BCL-2. HH signaling is normally silenced in adult cells but becomes activated and deregulated in myeloid malignancies, including MPN. Several HH inhibitors have been developed that target SMO, including GDC-0449 (Vismodegib), LDE225, and PF-04449913. Recently, a first-in-man, phase Ia dose escalation study was conducted with PF-04449913, an oral SMO antagonist HH inhibitor in patients with hematologic malignancies, including MF. Patients with MF treated with PF-04449913 attained stable disease, and 1 patient treated for more than 385 days, achieved a clinical improvement, with 50% reduction in spleen size that was sustained for more than 8 weeks. This study showed that PF-04449913 was safe to administer, was well tolerated, and displayed promising activity in treating MPN. These findings support the rationale to further test Hh signaling inhibitors alone and in combination with JAK-TKI against MPN cells.
β-Catenin Antagonist
The canonical WNT-β-catenin signaling is essential to the self-renewal and growth of myeloid leukemia stem and MPN progenitor cells. In transformed myeloid stem and progenitor cells with activating tyrosine kinase mutations, deregulated WNT signaling has been noted to inhibit polyubiquitylation and proteasomal degradation of β-catenin by the SCF-like complex comprising Siah-1, SIP, Skp1, and TBL1. The multiprotein degradation complex for β-catenin is often inactivated in transformed HPCs. This situation results in the preservation, nuclear translocation, and interaction of β-catenin with the T-cell factor (TCF)/lymphoid enhancer factor transcription factor, which regulates the expression of genes such as Cyclin D1, MYC, and Survivin. In a recent study, the activity of BC2059 (β-Cat Pharmaceuticals, Gaithersburg, MD), a potent small-molecule, anthraquinone oxime-analogue inhibitor of the WNT-β-catenin pathway, was determined in human cultured and primary MPN cells derived from patients with MF versus normal CD34+ bone marrow progenitor cells. Treatment with BC2059 mediates the degradation and attenuated levels of β-catenin. Exposure to 100 nM of BC2059 induced cell cycle G1 phase accumulation and apoptosis of the cultured MPN HEL92.1.7 (HEL) and UKE1 cells expressing the mutant JAK2V617F. BC2059 treatment also induced apoptosis of CD34+ primary MPN cells from patients with advanced MPN expressing mutant JAK2. In contrast, BC2059 did not induce significant apoptosis of normal CD34+ progenitor cells. Exposure to BC2059 attenuated both β-catenin protein levels (significantly restored by cotreatment with the proteasome inhibitor) and the activity of the LEF1/TCF4 transcription factor, associated with reduced levels of Cyclin D1, MYC, and survivin in the cell lysates of BC2059-treated MPN cells. In a xenograft model of HEL cells in NOD/SCID mice, treatment with BC2059 showed significantly improved survival ( P <.001). Compared with treatment with each agent alone, cotreatment with BC2059 (20–50 nM) and JAK2-TKI TG101209 (200–1000 nM) synergistically induced apoptosis of HEL and primary CD34+ MPN cells, but was remarkably less toxic against normal CD34+ progenitor cells ( P <.01). These preclinical studies highlight the therapeutic potential of β-catenin antagonists alone and in combination with JAK-TKI against MPN, which needs to be further explored clinically.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


