Chapter Outline
PATHOLOGIC CLASSIFICATION OF RHABDOMYOSARCOMA
RHABDOMYOSARCOMA IN CANCER PREDISPOSITION SYNDROMES
GENETICS OF SPORADIC RHABDOMYOSARCOMA
Gene Fusions Generated by Chromosomal Rearrangements
Chromosome Copy Number Changes
Amplification of Specific Chromosomal Regions
Oncogene and Tumor Suppressor Gene Mutations in Rhabdomyosarcoma
Hypermethylation as an Alternative Mechanism of Gene Inactivation in Rhabdomyosarcoma
APPLICATIONS OF MOLECULAR GENETIC APPROACHES TO DIAGNOSIS AND PROGNOSIS
Detection of Recurrent Gene Fusions
Pathologic and Clinical Differences between Fusion Subtypes
Gene Expression Profiling with Microarrays
Detection of Rhabdomyosarcoma in Sites Other Than Primary Tumor
MOLECULAR AND CELLULAR BIOLOGY OF RHABDOMYOSARCOMA
Myogenic Pathways in the Tumorigenesis of Rhabdomyosarcoma
Role of Insulin-like Growth Factors in Rhabdomyosarcoma
Modulation of Growth and Apoptotic Pathways by Fusion Oncoproteins
PRECLINICAL INVESTIGATION OF EMERGING DRUG TARGETS FOR RHABDOMYOSARCOMA
Preclinical Models to Study Rhabdomyosarcoma Tumorigenesis
Inhibitors of Receptor Tyrosine Kinases
Inhibitors of Intracellular Signaling Pathways
Inhibitors of Developmental Pathways
Inhibitors of Other Cellular Processes That Support Rhabdomyosarcoma Tumorigenesis
CLINICAL RESEARCH ON RHABDOMYOSARCOMA
CLINICAL PRESENTATION AND EVALUATION
Pathologic Classification of Rhabdomyosarcoma
The term rhabdomyosarcoma (RMS) comprises a heterogeneous family of soft tissue cancers that are related in poorly understood ways to the skeletal muscle lineage. Some of these tumors occur in the vicinity of skeletal muscle, but others occur in areas without obvious skeletal muscle; thus these cancers cannot be defined solely as tumors of skeletal muscle. Instead, this family is derived from unknown mesenchymal precursors and is characterized by a shared program of skeletal myogenesis. Despite this shared program, some precursors may intrinsically undergo myogenesis whereas others may have developed this ability aberrantly.
Histopathologic classification has evolved over time, but two principal histopathologic subtypes have been recognized consistently in children: alveolar RMS (ARMS) and embryonal RMS (ERMS). The criteria for these categories were refined over time as these subtypes were associated with clinically distinct phenotypes. As described in detail later, ARMS more often occurs in the extremities and axial musculature in older children and is associated with a less favorable prognosis whereas ERMS tends to occur in the head, neck, and genitourinary tract of younger patients and is associated with a favorable prognosis. ARMS and ERMS account for 20% to 30% and 70% to 80% of RMS cases, respectively.
The diagnosis of RMS is often difficult because of the paucity of features of striated muscle differentiation. A variety of pediatric solid tumors, including RMS, neuroblastoma, Ewing sarcoma, and non-Hodgkin lymphoma, can present as collections of poorly differentiated cells (small round blue cell tumors). To detect more subtle evidence of myogenic differentiation, immunohistochemical reagents are used to identify muscle-specific proteins, such as MYOD, myogenin, desmin, myoglobin, and skeletal muscle-specific actin. In addition, the detection of myofilaments by electron microscopy provides further evidence of the myogenic phenotype.
In ARMS, the tumor cells are small blue round cells with only small amounts of cytoplasm ( Fig. 59-1, A ). The prominent round nuclei generally have monotonous, coarse chromatin, and central nucleoli are often present. These tumor cells form highly cellular aggregates separated by fibrovascular septa. Within these aggregates, discohesive areas tend to form, resulting in spaces or clefts lined by rhabdomyoblasts. ARMS was named for these open spaces, which have an appearance reminiscent of pulmonary alveoli. In addition to these features, the tumor cells tend to align along septa in a “picket-fence” pattern and tumor giant cells are often present. In a subset of cases there is a paucity of fibrovascular stroma, no evidence of spaces, and the predominance of a highly cellular small round cell population with cytologic features similar to the classic alveolar pattern; such cases are referred to as the solid variant of ARMS.
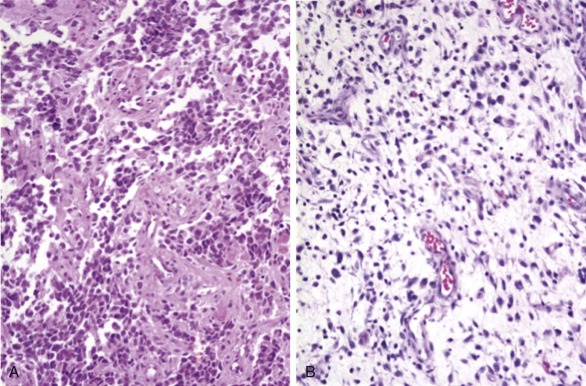
ERMS is so named because of its histologic similarity to developing skeletal muscle. The tumor cells show varying degrees of differentiation along the myogenic spectrum, from small primitive round cells to larger oblong cells with eccentric oval nuclei and varying amounts of eosinophilic cytoplasm (see Fig. 59-1, B ). ERMS nuclei are often notable for a relatively bland chromatin pattern. These differentiated cells can elongate to assume a straplike appearance and occasionally show cross-striations and multinucleation. In addition to the characteristic cytology, ERMS tumors classically have variable cellularity, in which areas of hypercellularity alternate with areas of hypocellularity in a loose myxoid stroma.
Within the ERMS category, there are several histologic variants. In sclerosing RMS, there is abundant desmoplasia and the tendency to form microalveolar tumor cell nests within the desmoplastic areas ; in some cases, this microalveolar appearance has been mistaken for ARMS. In a second variant, there are dense clusters of ERMS cells that can be confused with the solid variant of ARMS ; this dense ERMS variant can be distinguished based on cytologic features and other special studies. Finally, two variants of ERMS have been associated with an improved prognosis. Botryoid RMS typically occurs in the lumen of a hollow viscus, such as the urinary bladder, vagina, or extrahepatic bile ducts, and grossly has multiple polypoid nodules. At the microscopic level, this tumor typically has a dense cambium layer of tumor cells under an intact epithelial surface. Spindle-cell RMS has dense bundles or whorls of spindle-shaped cells that resemble smooth muscle. In children, these lesions commonly occur in the paratesticular region, but in adults, in whom the prognosis is worse, these lesions tend to occur in the head and neck and are often associated with a sclerosing pattern. In both of these latter two variants, the tumors cells can show marked rhabdomyoblastic differentiation.
Another small subset of RMS cases demonstrates discrete areas with ERMS-like histology and separate discrete areas with ARMS-like histology. The ERMS-like and ARMS-like areas can have any one of the various histologic patterns described earlier. Although these cases were previously diagnosed as ARMS or ERMS based on prevailing histopathologic criteria, these cases are currently classified as mixed RMS.
A final RMS subset shows features similar to anaplastic Wilms tumors. Although the anaplasia can be seen in both ARMS and ERMS, it is more prevalent in the latter. Anaplastic RMS tumors have large, lobated hyperchromatic nuclei and atypical mitoses. In some cases anaplastic cells are scattered in small patches (focal anaplasia), whereas other cases have larger clusters or sheets of anaplastic cells (diffuse anaplasia).
Rhabdomyosarcoma in Cancer Predisposition Syndromes
The majority of RMS cases arise as sporadic nonheritable tumors, but a small fraction of cases are associated with heritable genetic syndromes ( Table 59-1 ). In some cases the proband with RMS inherited a mutant gene as part of an established familial syndrome, and in other cases a new germline mutation occurred in one germ cell that ultimately produced the proband. The penetrance of RMS in individuals with new or inherited germline mutations varies from as high as approximately 7% in Costello syndrome to 0.1% to 1% in other syndromes. Although there are rare cases of RMS found in association with other syndromes, the determination that such syndromes increase predisposition to RMS is limited by the overall low frequency of these syndromes. In a similar way, the small contribution of known RMS susceptibility syndromes to the overall incidence of RMS is related to the low incidence of these syndromes in the general population. Neurofibromatosis type 1 has the highest prevalence, occurring in approximately 1 in 3500 individuals and accounting for about 1% of all RMS cases. Although TP53 germline mutations occur less frequently (~1 in 20,000 individuals), there is a higher prevalence of RMS in these mutation carriers; thus this susceptibility syndrome has been reported to occur in up to 10% all RMS cases.
| Cancer Syndrome | Locus | Gene | Nonneoplastic Findings | Characteristic Tumors | Frequency of RMS |
|---|---|---|---|---|---|
| Costello syndrome | 11p15.5 | HRAS | Facial anomalies, cardiac hypertrophy, cognitive defects, developmental delay | Benign: skin papillomas Malignant: RMS, neuroblastoma, bladder cancer | 7% |
| Li-Fraumeni syndrome | 17p13.1 22q12.1 | TP53 CHEK2 | None | Sarcoma, breast cancer, brain tumor, adrenocortical cancer, leukemia | 6% |
| Hereditary retinoblastoma | 13q14 | RB1 | None | Retinoblastoma, osteosarcoma | 2% |
| Neurofibromatosis type 1 | 17q11.2 | NF1 | Café au lait spots, skinfold freckling, Lisch nodules, cognitive deficits | Benign: neurofibroma Malignant: MPNST, AML | 1% |
| Constitutional mismatch-repair-deficiency syndrome | 7p22.2 and others | PMS2, MLH1, MSH2, MSH6 | Café au lait spots | hematologic cancers, brain tumors, colorectal cancer | 1% |
| Beckwith-Wiedemann syndrome | 11p15.5 | Unknown | Macrosomia, macroglossia, hemihyperplasia, visceromegaly | Wilms tumor, hepatoblastoma | <1% |
| Nevoid basal cell carcinoma syndrome | 9q22 | PTCH | Macrocephaly, skin cysts, palmar and plantar pits, rib anomalies | Basal cell carcinoma, medulloblastoma | <1% |
| Rubinstein-Taybi syndrome | 16p13.3 | CREBBP | Mental retardation, facial anomalies, and broad thumbs | Leukemia, brain tumors | <1% |
| Noonan syndrome | 12q24 and others | PTPN11, SOS1, RAF1, and others | short stature, facial anomalies, congenital heart defects | Neuroblastoma, ALL, glioma, RMS | <1% |
| DICER1 syndrome (formerly PPB family tumor and dysplasia syndrome) | 14q32 | DICER1 | None | PPB, cystic nephroma | Unknown |
Among the syndromes predisposing to RMS, three syndromes (termed RASopathies ) are caused by alterations of the RAS signaling pathway. Costello syndrome is caused by mutations in HRAS codons 12 or 13. These same mutations commonly occur in many types of sporadic tumors, have transforming activity in cell culture, and thus are gain-of-function alleles. Similarly, Noonan syndrome is caused by activating mutations of one of several oncogenes encoding downstream RAS pathway components with growth stimulatory function. In contrast, neurofibromatosis type 1 is caused by inactivating mutations of the NF1 tumor suppressor gene, which encodes neurofibromin, a guanosine triphosphatase–activating protein that negatively regulates RAS signaling. The presence of a common affected signaling pathway is also indicated by the significant overlap in nonneoplastic phenotype in both Costello and Noonan syndromes. In contrast, neurofibromatosis type 1 is associated with different nonneoplastic phenotypic features. Analysis of these heritable syndromes has thus shown that activation of RAS signaling by one of several genetic changes leads to a low and variable frequency of RMS susceptibility.
The RMS tumors associated with these hereditary predisposition syndromes display a number of notable properties. In many of these syndromes, the RMS tumors occur at a younger age than sporadic RMS tumors, which is in accord with the two-hit tumor susceptibility gene model. Of note, RMS cases in hereditary retinoblastoma occur at a later age but are associated with radiation treatment of the primary retinoblastoma, thus suggesting the need for a postnatal DNA damage event for RMS in this setting. From a pathology perspective, most of these syndromic RMS tumors are diagnosed as ERMS, the subtype that is not associated with the PAX3-FOXO1 and PAX7-FOXO1 gene fusions (described later). In the few cases in which tumors are described as ARMS, such as in hereditary retinoblastoma or Beckwith-Wiedemann syndrome, molecular testing reveals that these RMS tumors are fusion negative.
Genetics of Sporadic Rhabdomyosarcoma
Gene Fusions Generated by Chromosomal Rearrangements
Cytogenetic studies established that nonrandom chromosomal translocations distinguish most ARMS tumors from ERMS and other pediatric solid tumors ( Fig. 59-2 ). A translocation involving chromosome 2 and 13, t(2;13)(q35;q14), was found in 56% of 99 published ARMS cases, and a variant translocation involving chromosome 1 and 13, t(1;13)(p36;q14), was identified in 6% of cases ( http://cgap.nci.nih.gov.easyaccess2.lib.cuhk.edu.hk/Chromosomes/Mitelman ). This latter fraction is probably an underestimate because the 1;13 translocation is usually followed by an amplification event (see later). In contrast to ARMS, the 2;13 and 1;13 translocations were found in 3% and 0% of 77 published ERMS cases, respectively. Such rare translocation-positive ERMS cases may represent tumors with mixed histology or misdiagnosed ARMS tumors.
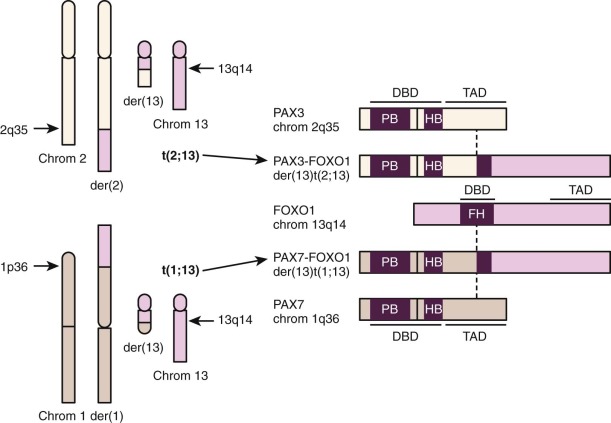
At the molecular level, the 2;13 and 1;13 translocations break and rejoin portions of transcription factor–encoding genes to generate fusion genes expressed as fusion transcripts (see Fig. 59-2 ). The rearranged genes on chromosomes 2 and 1 are PAX3 and PAX7, which encode related members of the paired box family. The chromosome 13 locus in these translocations is FOXO1 (FKHR), which encodes a member of the forkhead family. Although two reciprocal fusion genes, PAX3-FOXO1 and FOXO1-PAX3 (or PAX7-FOXO1 and FOXO1-PAX7 ), are formed by these translocations, the higher and more consistent expression of PAX3-FOXO1 or PAX7-FOXO1 supports the premise that the PAX3-FOXO1 or PAX7-FOXO1 products are involved in ARMS pathogenesis.
The PAX3-FOXO1 and PAX7-FOXO1 fusion transcripts are translated into novel chimeric transcription factors (see Fig. 59-2 ). The wild-type PAX3, PAX7, and FOXO1 genes encode transcription factors organized with an amino-terminal (N-terminal) DNA-binding domain and a carboxy-terminal (C-terminal) transcriptional activation domain. The translocations break within PAX3 or PAX7 intron 7 and maintain an intact DNA-binding domain but separate it from an essential part of the transactivation domain. In addition, the translocations break within FOXO1 intron 1 and disrupt the DNA-binding domain but maintain an intact transactivation domain. In the chimeric transcripts, the 5′ PAX3 (or 5′ PAX7 ) and 3′ FOXO1 coding sequences are fused in-frame to generate a 2508- or 2484-nucleotide open reading frame encoding an 836- or 828-amino acid fusion protein, respectively. These fusion proteins contain the first seven exons of PAX3 or PAX7, including an intact DNA-binding domain, and the last two exons of FOXO1, including an intact transactivation domain. A functional fusion protein cannot be created by other combination of PAX3 and FOXO1 exons because of incompatible reading frames or loss of functional domains. These rearrangements of PAX3 or PAX7 intron 7 and FOXO1 intron 1 thus appear to be selected because of functional constraints related to genomic organization of PAX3, PAX7, and FOXO1.
There are changes in both function and expression caused by the fusion events in ARMS. The chimeric transcription factors activate transcription from PAX3/PAX7-binding sites but are much more potent as transcriptional activators than the wild-type PAX3 and PAX7 proteins. This difference in transcriptional activity appears to be caused by a decreased sensitivity of the FOXO1 activation domain to the inhibitory effects of N-terminal PAX3 (or PAX7) domains, when compared with the effect of these N-terminal domains on the PAX3 (or PAX7) activation domain. The fusion proteins may also have a relaxed capability to bind and activate target genes owing to increased structural flexibility in the N-terminal DNA-binding domain. In addition to changes in transcriptional function, the PAX3-FOXO1 or PAX7-FOXO1 fusion products are expressed in ARMS tumors at higher levels compared with the corresponding wild-type PAX3 or PAX7 products. The gene-specific mechanisms for this enhanced fusion product expression are described later. Therefore these chromosomal changes result in high levels of chimeric transcription factors that inappropriately activate transcription of genes with PAX3/PAX7 DNA-binding sites. These biologic effects contribute to tumorigenesis by modulating myogenic differentiation, altering growth and apoptotic pathways, and stimulating motility and other metastatic pathways.
Several methodologies, including reverse-transcriptase polymerase chain reaction (RT-PCR) assay and fluorescent in situ hybridization (FISH), were applied to detect the PAX3-FOXO1 and PAX7-FOXO1 fusions in clinical material. A single large study of RMS cases from the Intergroup Rhabdomyosarcoma Study (IRS) IV protocol showed that all 93 cases of RMS or undifferentiated sarcoma with a diagnosis other than ARMS were fusion negative, whereas 77% of the 78 ARMS cases were fusion positive. Of these ARMS cases, 55% expressed PAX3-FOXO1, 22% expressed PAX7-FOXO1, and 23% were fusion negative. These findings were confirmed by several other large cooperative group studies. In other studies of mixed RMS, most but not all cases were fusion negative. Furthermore, when areas with ARMS-like and ERMS-like histology were individually examined by FISH, there was concordance for fusion status among the regions in a single tumor.
These molecular pathology studies establish that there is a significant subset (~20%) of ARMS cases that do not express either the PAX3-FOXO1 or PAX7-FOXO1 fusion. The IRS IV protocol and subsequent cooperative group studies used centralized pathology review and uniform tissue banking, and thus these fusion-negative results cannot be explained by inaccurate histopathologic diagnosis or suboptimal tissue samples. Subsequent analysis of these fusion-negative cases revealed that a small subset have variant fusions of PAX3, PAX7, or FOXO1 with other genes, including fusion of PAX3 with the FOXO1 -related locus FOXO4 (AFX1) and fusion of PAX3 with NCOA1 or NCOA2, which encode two similar transcription factors unrelated to the forkhead family. However, these variant fusions are uncommon; most fusion-negative ARMS cases do not have any rearrangements of PAX3, PAX7, or FOXO1 and thus appear to represent true fusion-negative cases with respect to these loci.
Although genetic studies have not revealed any chromosome rearrangements that occur in most ERMS cases, gene fusions have been identified in a small number of ERMS cases. In particular, rearrangements of the NCOA2 gene in the 8q11-13 chromosomal region were detected in four ERMS tumors. The pathologic subtype in three of the four cases was a spindle cell variant of ERMS. Furthermore, all four cases occurred in infants younger than 1 year of age and thus are characterized as congenital/infantile RMS. Further analysis of three cases demonstrated fusions to different partners in each case— PAX3 (2q35), SRF (8q11), and TEAD1 (11p15). Finally, there is a report of an unrelated fusion, EWSR1-DUX4, in an ERMS in a 19-year-old patient.
Chromosome Copy Number Changes
Various studies, including classic cytogenetics ( http://cgap.nci.nih.gov.easyaccess2.lib.cuhk.edu.hk/Chromosomes/Mitelman ), chromosome-based comparative genomic hybridization (CGH), and DNA-based microarrays established that a second set of differences between the RMS histopathologic subtypes is at the level of whole chromosome gains and losses. ERMS tumors have multiple numerical changes of whole chromosomes. The most frequent chromosome change in ERMS involves gains of chromosome 8. This change is characterized by polysomy, or multiple copies, of chromosome 8 and occurs in 50% to 90% of ERMS tumors but only 10% to 20% of ARMS tumors. This chromosome gain results in elevated expression of genes along chromosome 8 and cannot be localized to any specific chromosomal region. Additional common gains in ERMS involve chromosomes 2, 7, 11, 12, 13, and 20. In contrast to chromosome 8 gains, each of these latter changes is often a low-level gain, most likely a trisomy, and occurs in 20% to 50% of ERMS cases. Several of these gains, such as chromosome 2, 11, and 13, occur at significantly lower frequencies (2- to 10-fold) in ARMS tumors. Whole chromosome losses in ERMS commonly involve chromosomes 6, 9, 10, 14, 15, and 16 and occur in 15% to 25% of ERMS tumors. A study of intermediate-risk ERMS reports higher frequencies of these chromosome losses, suggesting that these events may vary among RMS risk groups. There are also quantitative differences in chromosome losses between ERMS and ARMS, but these differences are not as striking as the differences involving gains.
Although the chromosome gains and losses are not prevalent in the ARMS cases as a group, the fusion-negative ARMS subset has a pattern of gains and losses that is much more similar to ERMS than to the fusion-positive ARMS subset. The most striking finding involves chromosome 8; these events occur in more than 50% of fusion-negative ARMS tumors but not in fusion-positive ARMS tumors. As a second example, chromosome 11 gains occur in 30% of fusion-negative ARMS tumors but only 10% of fusion-positive ARMS tumors.
Amplification of Specific Chromosomal Regions
CGH and DNA-based microarray studies revealed that ARMS has significantly more genomic amplification events than ERMS. In CGH studies, one or more amplification events were detected in 47 of 84 ARMS cases (56%) compared with 7 of 43 ERMS cases (16%). The most common amplicons in ARMS tumors were derived from chromosomal regions 1p36, 2p24, 12q13-q14, 13q14, and 13q31 ( Table 59-2 ). The pattern of changes in the fusion-negative ARMS subset was again more similar to the pattern in ERMS than that in fusion-positive ARMS. This difference is exemplified by the finding of 12q13-q14 amplification in 24% of fusion-positive ARMS, 4% of fusion-negative ARMS, and 0% of ERMS. In addition to differences between fusion-positive and fusion-negative tumors, many of these high-frequency amplicons have a marked preference for either the PAX3-FOXO1– or PAX7-FOXO1– positive subset .
| Chromosome Region | Genes Involved | FREQUENCY | ||
|---|---|---|---|---|
| PAX3-FOXO1 | PAX7-FOXO1 | Fusion-negative | ||
| 1p36 | PAX7 | 0% | >90% | 0% |
| 2p24 | MYCN | 20% | 20% | 0-6% |
| 12q13-14 | CDK4 and others | 24 | <5% | <5% |
| 13q14 | FOXO1 | <10% | >90% | 0% |
| 13q31 | MIR17HG | <10% | 70% | <10% |
The amplification events at 1p36 and 13q14 generally occur in the same cases and are indicative of PAX7-FOXO1 amplification (see Table 59-2 ). In particular, PAX7-FOXO1 is amplified in more than 90% of PAX7-FOXO1– positive cases whereas PAX3-FOXO1 is amplified in less than 10% of PAX3-FOXO1– positive cases. As a further difference, there is a higher number of cells containing the amplicon in amplified PAX7-FOXO1– positive cases than in amplified PAX3-FOXO1– positive cases. At the expression level, the fusion transcript is expressed at higher levels in PAX7-FOXO1– positive cases than in PAX3-FOXO1– positive cases. Despite these differences, both fusion products are expressed in ARMS tumors at higher levels than the corresponding wild-type PAX3 or PAX7 products. This high expression is postulated to generate a fusion product level above a critical threshold for oncogenic activity. Although there is a common feature of fusion overexpression in fusion-positive ARMS, the mechanism of overexpression differs between the two subtypes. In PAX7-FOXO1 –positive tumors, the fusion is generally overexpressed because of genomic amplification, a copy number–dependent mechanism. In contrast, PAX3-FOXO1 is generally overexpressed by a copy number–independent increase in transcriptional rate.
For amplification of the 2p24 chromosomal region, the minimal amplified region contains the MYCN protooncogene (see Table 59-2 ). This genomic region corresponds to the same region that is frequently amplified in neuroblastoma. In ARMS there is a comparable incidence of 2p24 amplification (20%) in PAX3-FOXO1– and PAX7-FOXO1– positive tumors. In contrast, this amplification occurs very infrequently in ERMS and fusion-negative ARMS tumors (0% to 6% of cases). In addition to amplification, low copy number gains, which may be explained in part by whole chromosome 2 gains, were found in 50% to 60% of ARMS and ERMS cases. In fusion-positive tumors, 2p24 amplification generally results in high expression of the MYCN transcript and an increase in MYCN protein expression. The lower-level gains may also be associated with moderate increases in MYCN expression.
Amplification of the 12q13-q14 chromosomal region was found in numerous cancers, including bone and soft tissue sarcomas (e.g., osteosarcoma and liposarcoma), brain tumors (mostly glioblastoma), and carcinomas (including breast cancer). In ARMS, the minimal amplified region was localized to a 0.5-Mb region containing more than 20 genes, including CDK4, which encodes a cell cycle regulatory protein (see Table 59-2 ). The 12q13-q14 region is amplified preferentially in PAX3-FOXO1– positive cases; there is a 24% and 4% frequency of 12q13-q14 amplification in PAX3 – FOXO1– and PAX7-FOXO1– positive cases, respectively. This amplification also occurs at a low frequency in fusion-negative RMS tumors. In fusion-positive RMS tumors, 12q13-q14 amplification is associated with increased expression of multiple (but not all) genes in the minimal amplified region. Of note, the nearby 12q15 region (containing MDM2 ) is independently amplified at a low frequency in both fusion-positive and fusion-negative RMS tumors.
The 13q31 amplification event was localized to a 150-kb region surrounding the MIR17HG gene (see Table 59-2 ). This gene contains the miR-17-92 cluster, and the transcript expressed from the MIR17HG gene is processed to six microRNAs (miRs). This 13q31 region is also amplified in diffuse large B-cell lymphoma and small cell lung carcinoma. In RMS, this region is preferentially amplified in PAX7-FOXO1– positive cases; this amplicon is present in 70% of PAX7-FOXO1– positive cases and less than 10% of PAX3-FOXO1– positive RMS and fusion-negative RMS cases. In RMS tumors with 13q31 amplification, five of the six microRNAs in the cluster (miR-17, miR-19a, miR-19b, miR-20a, and miR-92a) are expressed at high levels. Transcripts from a number of tumor suppressor genes are targets of these microRNAs, and thus these microRNAs are postulated to be oncogenic by decreasing tumor suppressor expression.
Allelic Loss Events
RMS studies using polymorphic markers to distinguish between the two alleles at a locus reveal that allelic loss (or conversion to homozygosity) occurs commonly in ERMS tumors. With the use of DNA-based arrays with polymorphic markers, the genome-wide incidence of allelic loss (termed fractional allelic loss ) was twofold higher in ERMS compared with ARMS tumors. Furthermore, the fusion-negative ARMS subset demonstrated a fractional allelic loss similar to that of ERMS tumors that was more than twofold higher than that of fusion-positive ARMS tumors.
In ERMS, chromosome 11 is most commonly affected by allelic loss, and the smallest region of consistent allelic loss is 11p15.5 ( Fig. 59-3 ). This 11p15.5 allelic loss is also detected in a small subset (24%) of fusion-positive ARMS tumors but is much more common (77%) in ERMS and fusion-negative ARMS tumors. The probable presence of a tumor suppressor gene in this region, which is inactivated in RMS, is supported by several pieces of evidence. First, the gene or genes responsible for Beckwith-Wiedemann syndrome (which predisposes to several cancers, including RMS) were localized to this chromosomal region. Second, transfer of wild-type copies of chromosome 11 or fragments containing the 11p15 region suppresses growth of ERMS cells.
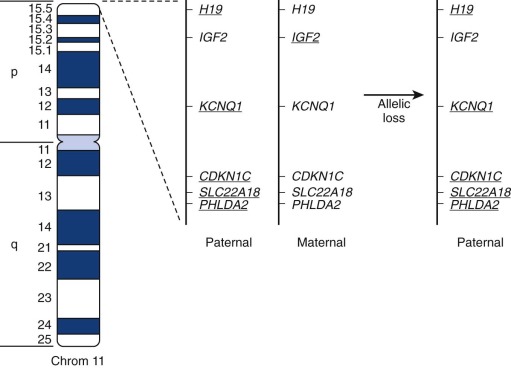
Determination of the parent of origin of the two alleles from the 11p15.5 region revealed that ERMS tumors preferentially maintain the paternal allele and lose the maternal allele (see Fig. 59-3 ). This preference suggests the action of genomic imprinting, a normal epigenetic developmental process that selectively inactivates expression of alleles in a gamete-of-origin–dependent process. Several genes in the human 11p15 chromosomal region and the corresponding mouse region show imprinting. For example, IGF2, encoding an embryonic growth factor, is preferentially expressed from the paternal allele, whereas CDKN1C, which encodes a cyclin-dependent kinase inhibitor (p57/KIP2) that negatively regulates cell-cycle progression, is preferentially expressed from the maternal allele. Two additional genes with preferential maternal expression and tumor suppressor function derive from opposite strands of the H19 locus, which produces the nontranslated H19 RNA, and the HOTS nucleolar protein. These combined studies of allelic loss in the 11p15.5 region suggest that ERMS tumorigenesis frequently involves inactivation of an imprinted tumor suppressor by allelic loss of the active maternal allele and retention of the inactive paternal allele.
Oncogene and Tumor Suppressor Gene Mutations in Rhabdomyosarcoma
The earlier-described studies of RMS patients with cancer predisposition syndromes revealed genes that are mutated and corresponding pathways that are altered in the pathogenesis of rare RMS cases. In particular, RMS cases occurred in syndromes in which the RAS pathway was activated whereas other cases occurred in syndromes in which the RB1 or TP53 signaling pathway was inactivated. The corresponding pathways thus are strong candidates for alterations in sporadic tumors, either by mutating the same genes that are altered in the predisposition syndromes or mutating other genes in these pathways.
Several studies suggest that small mutations are more common in ERMS than ARMS. In a recent next-generation sequencing (NGS) study of 147 RMS cases (53 fusion-positive and 94 fusion-negative), the mean rate of verified nonsynonymous mutations was fourfold higher in fusion-negative tumors (17.8 mutations/tumor) compared with fusion-positive tumors (6.4 mutations/tumor). Recurrent deleterious point mutations involving known genes were found in more than 50% of fusion-negative RMS cases compared with less than 5% of fusion-positive cases. Of note, only two recurrent point mutations ( PIK3CA and BCOR ) were found in the 48 fusion-positive ARMS cases whereas three recurrent point mutations were found in 11 fusion-negative ARMS cases. These overall findings in this NGS study were very similar to a second smaller NGS study as well as a directed mutation study. In this latter directed mutation study of 31 genes, mutations were detected in 14 of 57 (25%) ERMS cases compared with 1 of 21 (5%) ARMS cases. Except for one ERMS case with two mutations, the other 13 ERMS cases had only one mutation per case from this panel of 31 genes. An insufficient number of cases was examined to comment on mutation frequency in the fusion-negative ARMS subset.
The RAS pathway is frequently activated in sporadic ERMS tumors by mutations in one of several genes ( Table 59-3 ). In recent studies analyzing 147 ERMS cases, RAS family genes were mutated in 20% of ERMS cases. More than half of these RAS family mutations (19/31, 61%) occurred in the NRAS gene and the majority (15/19, 79%) of these NRAS mutations involved codon 61. In contrast, only one RAS family gene mutation (in KRAS ) was detected in 41 cases of ARMS (2%). Of various other RAS pathway components, PTPN11 and BRAF mutations were found in 3% and 1% of ERMS cases but not in any ARMS cases. In 1 of 12 ERMS cases (8%), a mutation was also found in the SPRY1 gene, which encodes a modulator of RAS signaling. Finally, homozygous deletions involving the NF1 locus were found in 15% of ERMS cases. The RAS , PTPN11, and BRAF point mutations as well as the NF1 deletions are mutually exclusive, consistent with the premise that these changes activate the same pathway. Therefore, one of several components in the RAS pathway can be altered to activate this pathway in more than 40% of sporadic ERMS cases but in very few (<5%) sporadic ARMS cases.
| Gene | ERMS | ARMS | Other † | All RMS |
|---|---|---|---|---|
| HRAS | 5/147 ‡ (3%) | 0/41 (0%) | NA | 5/188 (3%) |
| KRAS | 7/147 (5%) | 1/41 (2%) | NA | 8/188 (4%) |
| NRAS | 19/147 ‡ (13%) | 0/41 (0%) | NA | 19/188 (10%) |
| BRAF | 1/93 (1%) | 0/31 (0%) | 0/29 (0%) | 1/153 (1%) |
| MAP2K1 | 0/80 (0%) | 0/21 (0%) | NA | 0/101 (0%) |
| MAP2K2 | 0/23 (0%) | NA | NA | 0/23 (0%) |
| PTPN11 | 3/108 (3%) | 0/31 (0%) | NA | 3/139 (2%) |
| SPRY1 | 1/12 (8%) | 0/7 (0%) | NA | 1/19 (5%) |
| SOS1 | 0/20 (0%) | NA | NA | 0/20 (0%) |
| CDKN2A | 0/27 (0%) | 0/17 (0%) | 6/44 (14%) | 6/88 (7%) |
| CTNNB1 | 2/57 (4%) | 0/21 (0%) | NA | 2/78 (3%) |
| DICER1 | 4/57 (7%) | NA | NA | 4/57 (7%) |
| FGFR4 | 11/112 (10%) | 2/47 (4%) | 2/12 (17%) | 15/171 (9%) |
| PIK3CA | 3/57 (5%) | 0/21 (0%) | 1/12 (8%) | 4/90 (4%) |
| TP53 | 2/50 (4%) | 0/41 (0%) | 10/68 (15%) | 12/159 (8%) |
* RMS cell lines were not included in this compilation.
† Other includes Botryoid, pleomorphic, mixed, and unclassified cases.
‡ In one ERMS case, both a HRAS and a NRAS mutation were detected.
| Gene | ERMS | ARMS | Other † | All RMS |
|---|---|---|---|---|
| CDKN2A/B | 8/32 (25%) | 1/6 (17%) | 2/44 (5%) | 11/82 (13%) |
| NF1 | 4/26 (15%) | NA | NA | 4/26 (15%) |
| RB1 | 6/27 (22%) | 2/20 (10%) | NA | 8/47 (17%) |
| TP53 | 1/4 (25%) | 0/2 (0%) | 1/31 (3%) | 2/37 (5%) |
* RMS cell lines were not included in this compilation.
† Other includes Botryoid, pleomorphic, mixed, and unclassified cases.
Alterations in genes encoding RB1 or proteins regulating RB1 function occur in sporadic ERMS and ARMS tumors, providing several mechanisms to alter RB1 signaling. Although small RB1 mutations were not conclusively investigated in sporadic RMS, homozygous deletions were found in 22% (6/27) of ERMS and 10% (2/20) of ARMS cases (see Table 59-3 ). A higher frequency of RB1 gene alterations in ERMS is suggested by the finding of significantly lower RB1 expression in ERMS compared with ARMS tumors. As described earlier, 12q13-q14 amplification in a subset of ARMS cases results in overexpression of CDK4, which phosphorylates RB1 and contributes to RB1 functional inactivation. In another subset of RMS cases, homozygous deletions and/or point mutations occur in CDKN2A and CDKN2B, which encode inhibitors of CDK4 and CDK6. These alterations occur in both ARMS and ERMS, with an overall homozygous deletion frequency of 13% (11/82) and a mutation frequency of 7% (6/88).
There is also evidence for genetic changes that alter the TP53 pathway in sporadic RMS tumors (see Table 59-3 ). In multiple studies examining a total of 159 RMS cases, 12 missense mutations (8%) were detected in the TP53 gene. Although many cases were not subclassified for histopathologic subtype, mutations were found in both ARMS and ERMS. Homozygous deletions involving TP53 were also detected in a few cases. In addition to these direct changes in TP53, changes in the TP53 pathway may be caused by MDM2 gene amplification (described earlier), which encodes a protein that binds to and inactivates TP53. Finally, deletion of the CDKN2A locus (described earlier) also eliminates expression of p14ARF, which is a protein that normally blocks MDM2 function and stabilizes the TP53 protein.
Activation of several additional signaling pathways is also implicated in sporadic RMS tumors (see Table 59-3 ). Mutation in the tyrosine kinase domain of the membrane receptor fibroblast growth factor receptor 4 (FGFR4) was detected in 15 of 171 (9%) RMS tumors, including 11 in 112 (10%) ERMS and 2 in 47 (4%) ARMS cases. These mutations activate downstream signaling from FGFR4 and increase proliferation, invasion, and metastatic potential in mutation-bearing cells. Other genes in which mutations were found only in ERMS tumors include PIK3CA (3/57, 5%) and CTNNB1 (2/57, 4%); these mutations will activate phosphatidylinositol-3-kinase and Wnt signaling pathways, respectively. Although activation of the Hedgehog signaling pathway has been noted in a substantial subset of ERMS tumors, point mutations were not found in genes encoding several classic pathway components. As a possible alternative explanation of this increased Hedgehog signaling, low-level gains of the GLI1 locus were found in numerous ERMS cases. Finally, DICER1 mutations have been detected in 2 of 52 (4%) sporadic ERMS tumors and result in deregulated microRNA processing.
Hypermethylation as an Alternative Mechanism of Gene Inactivation in Rhabdomyosarcoma
In a final set of genomic changes in RMS, genetic loci can be silenced by epigenetic events involving tumor-specific hypermethylation of CpG islands. The cytosine residues in CpG dinucleotides are ordinarily unmethylated in normal cells but can become methylated during tumorigenesis, thereby altering the nearby chromatin structure and silencing transcription of associated genes. Studies of small numbers of RMS cases indicated that several genes often hypermethylated in other tumor types, such as CDKN2A, are not affected in a significant subset of RMS cases. However, hypermethylation of the CpG islands in the RASSF1A, HIC1, and CASP8 genes was detected in both ARMS and ERMS tumors. In contrast, there is evidence of differential methylation of the CpG islands in the MYOD and PAX3 genes such that hypermethylation occurs in most ERMS cases and hypomethylation occurs in most ARMS cases. A genome-wide approach also identified 140 promoter regions hypermethylated in at least four of ten RMS tumors compared with skeletal muscle, including genes implicated in development and tumorigenesis, such as GATA6 and HES5. Unsupervised hierarchical cluster analysis indicates that DNA methylation pattern correlates with RMS subtype. These hypermethylation events are generally associated with loss of expression of the corresponding proteins, providing an alternative nonmutagenic mechanism for inactivating genes and altering associated pathways.
Applications of Molecular Genetic Approaches to Diagnosis and Prognosis
Detection of Recurrent Gene Fusions
As described earlier, RT-PCR and FISH methodologies were developed to detect the PAX3-FOXO1 and PAX7-FOXO1 gene fusions in clinical material. In contrast to Southern blot approaches, both RT-PCR and FISH can be applied to detect these fusions in small amounts of input tissue. Furthermore, both RT-PCR and FISH approaches have been developed for formalin-fixed paraffin-embedded tissues as well as fresh and frozen tissues. Another favorable feature of the RT-PCR assays is high technical sensitivity, which permits detection of micrometastatic spread of rare fusion-positive cells to tissues such as bone marrow. Although FISH has lower technical sensitivity, a break-apart FOXO1 FISH assay has simplified detection of FOXO1 rearrangements resulting from either PAX3-FOXO1 or PAX7-FOXO1. In addition, break-apart PAX3 and PAX7 assays facilitate detection of variant fusions in ARMS cases that do not contain either of the two usual fusions.
Pathologic and Clinical Differences between Fusion Subtypes
Clinical-molecular correlative studies revealed several differences between fusion-negative and fusion-positive tumors. In comparisons of outcome among the fusion subsets, the fusion-negative ARMS cases have a failure-free survival (FFS) and overall survival (OS) similar to ERMS cases and significantly better than fusion-positive ARMS cases. There is a similar frequency of other parameters, such as unfavorable sites and metastasis, in ERMS and fusion-negative ARMS, and this frequency is lower in fusion-negative ARMS than in fusion-positive ARMS. Finally, in an examination of histopathologic parameters, nearly half of fusion-negative cases lack cystic foci and show totally “solid alveolar” architecture.
Although there are no detectable histopathologic differences between PAX3-FOXO1– and PAX7-FOXO1– positive cases, there are clinical differences between these two fusion-positive subsets. The OS rate in the PAX7-FOXO1– positive subset is superior to that in the PAX3-FOXO1– positive subset and similar to the OS rate in the ERMS and fusion-negative ARMS categories. In contrast, failure free survival in the two fusion-positive subsets is similar. In addition to OS, there are other clinical differences between these two fusion-positive subsets. In particular, the PAX7-FOXO1– positive tumors are typically diagnosed at a younger age, more often present with an extremity primary tumor, and are associated with a lower frequency of local invasion and nodal metastasis. A multivariate analysis of the contributions of prognostic parameters found that fusion status was an independent predictor of OS. A risk-based stratification incorporating fusion status, stage, and age has potential utility for stratifying the Children’s Oncology Group (COG) intermediate-risk group into distinct subgroups.
Gene Expression Profiling with Microarrays
Microarray-based analyses of genome-wide gene expression compared RMS with other pediatric tumors included in the differential diagnosis of small blue round cell tumors. Cases were clustered based on differences and similarities in expression patterns. Statistical analyses identified lists ranging from 20 to 93 genes that can correctly classify all tumors. These lists contain genes specifically expressed in each tumor category, including muscle-specific and myogenesis-related genes that are specifically expressed in RMS. With the use of minimization strategies, the criteria needed for classification were reduced in size to lists ranging from 7 to 31 genes. To extend these findings, 39 genes were selected to design a multiplex RT-PCR assay, which generates findings comparable to the microarray platform.
Microarray studies also developed classifiers for subsets within the RMS family. With the use of clustering algorithms, it was shown that the PAX3-FOXO1– and PAX7-FOXO1– positive subsets of ARMS cluster together, indicating no major expression differences between the two fusion-positive subsets. In contrast, there was a striking difference in expression pattern between the fusion-positive and fusion-negative ARMS cases. There were four published cases with variant fusions: two clustered with fusion-positive tumors and two clustered with the fusion-negative tumors. These analyses also demonstrated a striking difference in expression pattern between ERMS and fusion-positive ARMS cases. Comparison of ERMS and fusion-negative ARMS cases did not reveal any recurrent expression differences between these two groups of fusion-negative RMS cases. Therefore, based on expression pattern, RMS cases can be divided into two major categories: the fusion-positive and fusion-negative cases. Furthermore, when viewed on a multidimensional scaling plot, the fusion-positive group showed a dense cluster whereas the fusion-negative group showed a cluster with more heterogeneous spread.
The fusion-negative RMS category is proposed to be further divisible into three novel subsets, which are related to expression of muscle differentiation and chromosome 11 genes. A well-differentiated RMS subset expressed both gene sets, whereas a moderately differentiated RMS subset expressed the chromosome 11 genes with decreased levels of muscle differentiation genes. A final subset containing approximately 25% of ERMS tumors as well as undifferentiated sarcomas and non-RMS soft tissue tumors (NRSTS) did not express either gene set. In addition, tumors in this last subset had a fractional allelic loss (including 11p loss) similar to that in fusion-positive RMS tumors and twofold lower than that in the well-differentiated and moderately differentiated subsets. The significance of these subsets is highlighted by the finding of good, intermediate, and poor outcomes in well-differentiated, moderately differentiated, and undifferentiated sarcoma/NRSTS categories, respectively.
For prospective diagnosis of fusion-positive or fusion-negative RMS, genes differentially expressed between the two fusion subsets were identified to generate initial signatures ranging from 121 to 534 genes. The genes preferentially expressed in fusion-positive tumors were overrepresented in PAX3-FOXO1 downstream targets (including genes involved in neurogenesis) and chromosome 6 genes, whereas the genes preferentially expressed in fusion-negative tumors were overrepresented in chromosome 8 genes. Additional statistical approaches identified minimal signatures consisting of 10, 5, or even 2 genes with predictive accuracy of 95% or greater. To develop an assay applicable to formalin-fixed paraffin embedded samples, biomarkers were identified that correspond to differentially expressed genes with commercially available antibodies suitable for immunohistochemistry. These efforts identified epidermal growth factor receptor (EGFR), fibrillin-2, and high-mobility group AT-hook 2 (HMGA2) as markers expressed in fusion-negative RMS cases and transcription factor AP-2β (TFAP2B) and P-cadherin as markers expressed in fusion-positive RMS cases. The combination of two markers for each RMS subset achieved specificity of 90% or greater and a sensitivity of 60% or greater.
Several microarray and other genomic approaches were used to identify fusion protein downstream targets. In one approach, transduction of PAX3-FOXO1 or PAX7-FOXO1 in an ERMS cell line modulated expression of 334 genes (266 increased and 68 decreased); 81 of these genes (24%) were also differentially expressed between fusion-positive and fusion-negative RMS tumors. In the second approach, siRNA-mediated downregulation of PAX3-FOXO1 in an ARMS cell line decreased expression of 1834 genes after 24 hours of treatment; only 51 (3%) of these genes overlapped the differential expression signature in RMS tumors. In a final approach, ChIP-Seq analysis identified 1463 genomic regions bound to the fusion protein in ARMS cells; these genomic regions correspond to 24% of genes expressed at higher levels in fusion-positive RMS and 6% of the genes expressed at higher levels in fusion-negative RMS. These findings indicate that only a fraction of expression differences between fusion-positive and fusion-negative tumors is directly attributable to the fusion protein; many of the expression differences may result from other genetic and environmental differences between these RMS subsets.
Microarray analysis also elucidated genes whose expression predicts outcome in RMS patients. In particular, Cox regression proportional hazards modeling of OS with RMS microarray expression data identified 15 or 34 gene sets whose aggregate pattern of expression was correlated with OS. The performance of these two gene sets was high in the initial patient cohort and lower but still significant in a validation cohort. For the 34 gene set, this expression-based stratification of RMS cases was related to the current COG risk classification system. In addition, this expression-based stratification for both the 15- and 34-gene sets was also correlated with PAX3-FOXO1 and PAX7-FOXO1 fusion status. Multivariate analysis showed that these 15- or 34-gene sets were not independent predictors of outcome.
A hypothesis was also proposed that fusion protein downstream targets may predict outcome. Starting with 81 target genes that were both modulated by exogenous fusion protein in ERMS cells and differentially expressed between the fusion-positive and fusion-negative tumors, Cox regression modeling elucidated a 28-gene predictor. An aggregate score calculated from expression of these 28 genes is predictive of OS in ARMS and independent of known prognostic variables. This finding suggests that modulation of downstream fusion protein target expression is associated with differences in biologic aggressiveness of ARMS.
Detection of Rhabdomyosarcoma in Sites Other Than Primary Tumor
Molecular markers in conjunction with high-sensitivity detection methodologies have been applied to screen for RMS dissemination in accessible and commonly involved sites such as bone marrow, peripheral blood, and lymph nodes ( Table 59-4 ). In general, these approaches screen for molecular markers within cells to identify cases that are not detected by conventional radiology or pathologic examination. One strategy for minimal disseminated disease detection focuses on tumor-specific markers, such as PAX3-FOXO1 and PAX7-FOXO1. A second strategy, which is broadly applicable to all RMS subtypes, assays lineage-specific markers, such as myogenic markers of determination and differentiation (including MYOD1 and MYOG). In a third strategy, sites of disease spread were assayed for proteins involved in metastatic pathways, such as CXCR4 and MET. RT-PCR assays were developed to detect several of these markers and demonstrate analytical sensitivity in the range of one positive cell per 10 4 to 10 5 normal cells. More recent PCR protocols use real-time technology to quantify the extent of disseminated disease and can thus monitor changes in disease load over time and compare results to an empirically determined clinical threshold. In addition to PCR assays, antibodies have been used to detect various protein markers in immunocytochemical or flow cytometry assays. These assays also have a high level of analytical sensitivity (~10 −4 ), which is significantly better than conventional morphology-based evaluation.
| Marker | Molecule | Target | Assay |
|---|---|---|---|
| ACHRA | mRNA | Tumor cells | RT-PCR |
| ACHRG | mRNA | Tumor cells | RT-PCR |
| PAX3/PAX7-FOXO1 | mRNA | Tumor cells | RT-PCR |
| MYOD1 | mRNA protein | Tumor cells | RT-PCR Immunocytology |
| MYOG | mRNA protein | Tumor cells | RT-PCR Immunocytology |
| CXCR4 | Protein | Tumor cells | Immunocytology |
| MET | Protein | Tumor cells | Immunocytology |
| CD45/CD56 | Protein | Tumor cells | Flow cytometry |
| miR-206 | miRNA | Plasma/serum | RT-PCR |
| IGFBP2 | Protein | Plasma/serum | ELISA |
| VEGF-A | Protein | Plasma/serum | ELISA |
These minimal disseminated disease assays were applied to clinical material from RMS patients in several different settings (see Table 59-4 ). Multiple studies addressed the feasibility of detecting occult disease in bone marrow. These studies showed that histologically positive bone marrows are consistently detected, regardless of the methodology or marker. For histologically negative marrows, various methodologies and markers detected submicroscopic disease, with the frequency tending to be higher for ARMS relative to ERMS. In ARMS, which often metastasizes to the bone marrow, the frequency of a positive result in a histologically negative marrow ranges from 15% to 60%. In ERMS, in which bone marrow is not a common site of metastasis, the frequency of a positive result in a histologically negative marrow ranges from 7% to 33%. The presence of these marrow-infiltrating RMS cells is not consistently associated with subsequent overt marrow involvement, suggesting that the marrow can act as a transitory reservoir for disseminated RMS cells. Despite this finding, a significant association was found between poor outcome and detection of RMS cells in bone marrow; marrow involvement was associated with a significantly higher risk for recurrence (50% vs. 11%, P = .011) and poorer OS rate (47% vs. 92%, P =.01). In addition to bone marrow, studies also evaluated peripheral blood samples at diagnosis, during treatment, and after treatment. Comparison with outcome revealed that a positive result at the end of treatment was associated with poor outcome, and persistent positive results preceded a metastatic relapse.
Recent studies also examined expression of molecular markers in the serum or plasma. These markers include miR-206 and other muscle-specific miRNAs (see section below on microRNAs), which were measured by RT-PCR, and RMS-associated proteins, such as insulin-like growth factor–binding protein 2 (IGFBP2) and vascular endothelial growth factor (VEGF)- A (VEGF-A), which were assessed by enzyme-linked immunosorbent assay (ELISA). The finding of higher abundance of these markers in the plasma or serum of RMS patients relative to normal controls indicates a possible use in noninvasive diagnosis. Furthermore, an application in disease monitoring is suggested by the finding of decreased levels after treatment. Finally, an association of plasma IGFBP2 levels with metastatic RMS suggests a potential role in patient stratification for risk-based therapy.
Molecular and Cellular Biology of Rhabdomyosarcoma
Myogenic Pathways in the Tumorigenesis of Rhabdomyosarcoma
Based on the premise that RMS is related to the skeletal muscle lineage, the expression pattern of muscle-specific proteins has been extensively examined in RMS. In particular, many studies have focused on the family of myogenic transcription factors (MyoD, Myf5, myogenin, and MRF4) that are responsible in part for the determination of stem cells into myoblasts and then differentiation into myocytes. Assays of the corresponding genes detected RNA expression of MYOD and MYF6 in all ARMS and ERMS tumors and RNA expression of MYOG (myogenin) and MYF5 in all ARMS and most ERMS tumors. Antibodies directed to MyoD and myogenin have proved to be suitable for immunohistochemistry on paraffin-embedded, formalin-fixed tissues. With the use of these antibodies the vast majority of RMS cases (about 97% in a study of 956 cases) showed positive nuclear immunostaining for each protein. Because positive staining also occurred in multiple cases of pleuropulmonary blastoma, the specificity of these reagents was estimated to be between 90% and 91%. There are differences between ARMS and ERMS tumors in the myogenin staining patterns such that most cells within an ARMS tumor stain positive, whereas fewer cells within an ERMS tumor stain positive. This differential staining between ARMS and ERMS has also been shown for MyoD. There is a statistical difference in the extent of MyoD or myogenin expression among the RMS subtypes, but there is still overlap. Hence the immunohistochemical pattern of these myogenic proteins is not sufficient to classify cases but may be helpful in selecting cases for further testing.
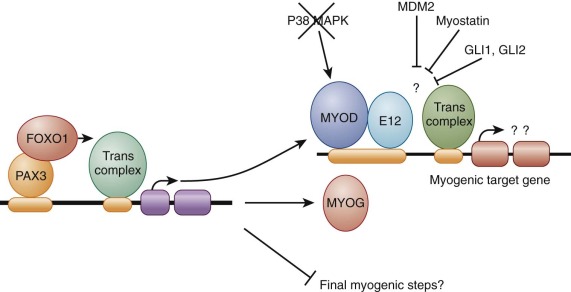
Functional studies in RMS cell lines determined that MyoD is not an active transcription factor in the RMS environment ( Fig. 59-4 ). In particular, although MyoD is able to bind DNA at its specific binding sites in RMS cells it is not capable of acting as a transcriptional activator on model genes with these DNA-binding sites. These effects may contribute to the failure of RMS cells to undergo terminal differentiation. Various factors may contribute to this inhibition of transcriptional activation by MyoD. Experiments fusing RMS cell lines to a second normal cell line resulted in some instances in which the transcriptional activation block was overcome, suggesting that the RMS line was deficient in a trans -acting factor. In other instances, the block was not overcome by this cell fusion, suggesting a dominant-acting inhibitory factor. As an example of the former recessive scenario, the p38 mitogen-activated protein kinase (MAPK) pathway is deficient in some RMS cell lines, resulting in the absence of an essential activator of MyoD during myogenic differentiation. Examples of the latter dominant scenario include amplification of MDM2 in an RMS cell line that inhibited MyoD activity by a mechanism involving competition for the transcription factor Sp1 and upregulation of musculin and a novel splice variant of E2A that inhibited MyoD activity by competing for heterodimer formation and preventing formation of functional MyoD:E-protein heterodimers, required for the switch to differentiation. In addition, increased expression of the negative muscle growth regulator myostatin, which is found in both ERMS and ARMS, is also capable of inhibiting MyoD-mediated transcriptional activity, possibly via Smad3 interactions. In RMS associated with activation of the Hedgehog pathway, expression of the transcription factor GLI1 or GLI2 inhibits MyoD-mediated transcriptional activation by reducing heterodimerization with its partner E12 and DNA binding. Finally, as discussed in the later section “Epigenetic Regulation,” MyoD activity can also be inhibited by decreased expression associated with KMT1A methyltransferase activity.
The identification of PAX3-FOXO1 and PAX7-FOXO1 gene fusions in ARMS also highlight PAX3 and PAX7 as important proteins in normal and aberrant myogenic transcriptional control. In murine systems, both Pax3 and Pax7 are involved in early embryonic development of cells giving rise to axial musculature, and Pax3 also has a necessary role in the early embryonic development of the limb musculature. In the adult mouse, Pax7 has an important role in the development of the major population of myogenic satellite cells and Pax3 is expressed in satellite cells in a subset of muscles. This developmental biology of PAX3 and PAX7 is relevant to ARMS tumors, which express PAX3 or PAX7 as a fusion with FOXO1, and to ERMS tumors, which express wild-type PAX3, PAX7, or both.
The wild-type and fusion PAX proteins can variably affect the myogenic status of the cells expressing them. When wild-type Pax3 is introduced into explanted embryonic tissues, it can activate the myogenic program as shown by expression of MyoD, Myf5, and myogenin as well as myogenic differentiation products. In contrast, Pax3 could not induce this myogenic program in NIH 3T3 murine fibroblasts, suggesting that this murine fibroblast represents a less permissive environment. Studies of the PAX3-FOXO1 fusion protein have demonstrated a range of activities on the myogenic program (see Fig. 59-4 ). First, studies focusing on the MyoD and myogenin promoters provide evidence that both of these genes are transcriptional targets of PAX3-FOXO1. Of two studies of NIH 3T3 cells, one indicated that PAX3-FOXO1 induced the myogenic program, including the upstream products MyoD, myogenin, Six1, and Slug as well as downstream myogenic products, such as troponins and myosin light chain. However, a second study of NIH 3T3 cells demonstrated induction of the upstream products MyoD, myogenin, and desmin but no induction of the downstream product myosin heavy chain. In contrast to these findings, transduction of PAX3-FOXO1 into two additional murine fibroblast lines (10T1/2 and Plus) resulted in expression of both upstream and downstream myogenic products and fusion into multinucleated myotubes. In contrast, introduction of PAX3 or PAX3-FOXO1 into two myogenic cell lines, C2C12 myoblasts and MyoD-expressing 10T1/2 cells, inhibited terminal myogenic differentiation after stimulation of differentiation by growth factor withdrawal. PAX3-FOXO1 was more potent than wild-type PAX3 in this phenotypic activity. Finally, repression of PAX3-FOXO1 expression in human ARMS cell lines with RNA interference (small interfering RNA [siRNA] or short hairpin RNA [shRNA] constructs) results in decreased proliferation without apparent apoptosis, decreased motility and invasion, and increased differentiation as demonstrated by morphologic and myogenic gene and protein expression. This finding indicates that PAX3-FOXO1 normally represses expression of this myogenic pathway in this cell type.
The finding of both stimulatory and inhibitory effects of PAX proteins on myogenic events may be explained by the hypothesis that these wild-type or fusion PAX proteins facilitate entry into the myogenic pathway and variably inhibit the final steps of the pathway depending on the cellular environment. In support of this hypothesis, data suggest that PAX3-FOXO1, by inducing MyoD expression but attenuating its activity, and inducing FGFR expression to block differentiation, simultaneously stimulates but blocks completion of myogenesis. This intermediate commitment to differentiation may be partially explained by recent studies in which expression of PAX3-FOXO1 in muscle satellite cells did not inhibit expression of MyoD but did inhibit expression of MyoD target genes.
MicroRNAs
Although decades of study shed light on the intrinsic cellular signaling pathways and extrinsic microenvironmental stimuli that control skeletal muscle differentiation during embryogenesis, postnatal life, and muscle repair, it was not until 2005 that another layer of control was identified. In work examining the spatiotemporal control of cardiogenesis, it was discovered that specific microRNAs were expressed in cardiac and skeletal muscle and served to balance proliferation and differentiation of muscle precursor cells. Since that time, a great deal of information on these small noncoding RNAs that enable the regulation of protein dosage through binding to 3′ ends of mRNA and affecting stability or translation to protein has been gathered and understood not only in the framework of normal skeletal myogenesis but also in tumors related to the skeletal muscle lineage such as RMS.
MicroRNAs influencing skeletal myogenesis can generally be divided into myomirs (miRs specific to skeletal and cardiac muscle) and nonmyomirs (miRs expressed in nonmuscle tissue but that can also be found in muscle.) The myomirs (nomenclature miR-1 to miR-206, which includes the miR-133 group) are encoded in bicistronic clusters on chromosomes 6, 18, and 20 and control such diverse developmental processes as somite organization, muscle precursor cell migration, survival, differentiation, fusion, and cross talk with epigenetic processes that control differentiation (see next section). Similar to other human cancers, RMS tumors are noted to have lost expression of miRs and their tumor suppressive properties. When miR-206 is expressed in human RMS cells, xenograft growth is halted and accompanied by differentiation, presumably by the orchestrated downregulation of hundreds of miR-206 target genes, including MET. Mir-206 expression levels also correlate with clinical behavior of RMS. Regarding the nonmyomirs, the reader is referred to two reviews on microRNAs and their role in RMS. For example, miR-185 suppresses RMS tumor growth and progression by targeting the Six1 oncogene (see later discussion under “ Metastatic Pathways in Rhabdomyosarcoma ”).
Finally, as was discussed in the section on “Rhabdomyosarcoma in Cancer Predisposition Syndromes,” germline or somatic mutations of DICER1, the enzyme that cleaves pre-microRNA to mature microRNA, have been found associated with ERMS. To date these mutations appear to be in the RNase IIIb domain, resulting in a decrease in its activity.
Epigenetic Regulation
Although the block to myogenic differentiation in RMS cells has been apparent for decades, only recently is there insight into some of the epigenetic changes enforcing this inhibition. As mentioned earlier, whereas the muscle-specific transcription factor MyoD can bind DNA in RMS, some targets fail to activate. For example, although MyoD binds similarly at the genome-wide level in both normal and RMS (in this case the human embryonal RMS cell line RD was studied), in RMS there is a subset of genes to which MyoD binds poorly and thus does not activate. This subset includes the transcription factors MEF2C, RUNX1, JDP2, and NFIC, found expressed at lower levels in RMS compared with normal muscle cells. There was also differential DNA accessibility, including changes in DNA methylation status. Therefore it has become clear that epigenetic changes contribute as much to RMS as changes in the DNA sequence itself. Below are summarized some of the epigenetic changes found in RMS, including changes in DNA methylation (introduced earlier in “Hypermethylation as an Alternative Mechanism of Gene Inactivation in Rhabdomyosarcoma”), changes in histone methylation, and changes in nucleosome positioning.
Methylation of gene promoters at CpG islands by DNA-specific methyltransferases serves to silence expression of target genes. Thus it would follow that tumors should demonstrate different DNA methylation profiles compared with nonmalignant tissue, and indeed a study of the genomes of RMS compared with normal skeletal muscle showed this to be the case. In both ERMS and ARMS, genes involved in development and differentiation were hypermethylated compared with normal skeletal muscle. However, there were also differences between ERMS and ARMS, with ARMS showing more hypermethylation of Polycomb target genes (see later.) As methods to improve the speed and resolution of the detection of DNA methylation patterns improve, further changes are expected to be identified.
Methylation of lysine residues in histones by Polycomb group remodeling complexes (known as PRC1 and PRC2) also serves to silence chromatin, by influencing nucleosome formation and function. Although this process occurs in a carefully controlled manner during development, if Polycomb complexes are inappropriately recruited to target genes or inappropriately activated due to component mutation, impaired commitment to cell lineage or a frank block in differentiation can ensue, resulting in a primitive cellular phenotype. For example, in both ERMS and ARMS the NF-κB-YY1-mir-29 regulatory circuit that ordinarily controls skeletal myogenesis is usurped; YY1 recruits PRC2 to silence target genes including MIR29 , so that muscle differentiation cannot occur. In ARMS, PAX3-FOXO1 transcriptionally activates JARID2 expression, which recruits PRC2 to the promoter regions of target genes including MYOG and MYL1 , again impairing differentiation. Recently, there has been some focus on the catalytic component of PRC2 known as the EZH2 methyltransferase. EZH2 was found upregulated at the protein level in RMS tumors ; and in loss-of-function studies in RD cells, silencing of EZH2-induced MyoD activation and partial reversal of the tumorigenic phenotype.
There are also methyltransferases independent of Polycomb group remodeling complexes that appear dysregulated in RMS. For example, the histone methyltransferase KMT1A (formerly known as Suv39h1), which methylates histone H3 on the lysine 9 residue, represses myogenic gene expression directed by MyoD. In ARMS this keeps the cells in an undifferentiated state. In support of its presumed oncogenic role, suppression of KMT1A by siRNA decreases ARMS cell and xenograft growth. In independent studies, a screen for tumor suppressors in a Kras- driven model of zebrafish ERMS revealed that point mutations in KMT1A suppress RMS formation.
Regarding the epigenetic changes that occur in response to nucleosome repositioning, components of the SWI/SNF chromatin remodeling tumor suppressor complex have been found mutated in RMS. An early hint of the involvement of the SWI/SNF complex was in the study of TPA-mediated differentiation of RD cells, which was shown to work by recruiting proteins including BRG1 (a subunit of SWI-SNF) to the myogenin promoter, driving differentiation. Later work showed that the BAF53a subunit of SWI/SNF is not downregulated in RMS and thus contributes to a differentiation block.
Role of Insulin-like Growth Factors in Rhabdomyosarcoma
Insulin-like growth factor 2 (IGF-2) is an important growth factor during the fetal period and is highly expressed in fetal skeletal muscle but not detectably expressed in adult skeletal muscle. High-level IGF-2 expression in murine myoblasts results in an increased proliferative rate, impaired myogenic differentiation, and anchorage independence, indicating a potential role of IGF-2 in neoplasia of the myogenic lineage. In accord with these findings, IGF-2 is highly expressed by both ERMS and ARMS tumors as well as in derived RMS cell lines ( Fig. 59-5 ). In addition to the growth factor, both RMS subtypes express the IGF-1 receptor (IGF-1R), which is a cell surface receptor for IGF-2 as well as IGF-1. This simultaneous expression of growth factor and receptor creates an autocrine loop that stimulates the growth and motility of RMS cells. No mutations have been detected in these genes; hence this autocrine situation may reflect the fetal muscle expression pattern that is maintained or induced by the other genetic alterations. The growth response is mediated through IGF-1R, whereas the motility response is mediated through the distinct IGF-2/mannose-6-phosphate receptor. Numerous studies have explored different strategies for interfering with the action of IGF-1R. These strategies include an antibody directed to this receptor, a kinase-deficient mutant that acts as a dominant negative, and an antisense construct directed to this receptor. Each of these strategies decreases the response of the cells to IGF-2 stimulation, inhibits cell growth and anchorage independence in culture, and inhibits tumorigenicity in vivo. Exploitation of this downregulation for preclinical and clinical studies is described in “ Preclinical Investigation of Emerging Drug Targets for Rhabdomyosarcoma ” and “ Clinical Research in Rhabdomyosarcoma ”.
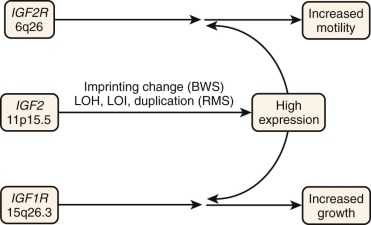
As described earlier, the IGF2 gene is located in the 11p15.5 chromosomal region, which is implicated in allelic loss events that occur in RMS tumors and in genetic alterations in the inherited disorder BWS, which predisposes to malignant tumors, including RMS. The IGF2 gene is part of an imprinted region and is preferentially expressed from the paternally inherited alleles. The result of the allelic loss events and genetic changes in BWS is loss of the nonexpressed maternal allele and maintenance or gain of the expressed paternal allele. In addition to these genetic changes, other alterations of 11p15 involving the IGF2 locus have been identified in both ARMS and ERMS tumors. In one study of 11 RMS tumors (including 4 ARMS tumors, 6 ERMS tumors, and 1 RMS tumor of uncertain classification), 1 ARMS and 3 ERMS cases demonstrated evidence of duplication of an IGF2 allele. Furthermore, although IGF2 is normally expressed from only one allele, loss of imprinting can occur and result in a biallelic expression pattern. In RMS, loss of imprinting at the IGF2 locus has been observed in 5 of 12 ERMS and 9 of 16 ARMS cases. These duplication or imprinting changes increase the number of active IGF2 alleles and may thereby contribute to the high expression of IGF2 mRNA and protein found in RMS tumors.
In mammalian cells, the subcellular localization of the wild-type FOXO1 (previously FKHR) and related FOXO3 and FOXO4 proteins is regulated by a PI3K-AKT (PKB-dependent) signaling pathway that is activated by survival- and growth-related signals such as IGF-2. In this signaling pathway, AKT-mediated phosphorylation of FOXO proteins leads to protein transfer from the nucleus to the cytoplasm and thus inactivates transcriptional function. Of the three AKT phosphorylation sites in FOXO1, two are retained in PAX3-FOXO1 and PAX7-FOXO1. If the fusion proteins are regulated by this pathway, the high level of IGF-2 in ARMS cells would cause these proteins to be phosphorylated and sequestered in the cytoplasm. However, experiments show that PAX3-FOXO1 is retained in the nucleus and is transcriptionally active in cultured cells in the presence of active AKT and in ARMS cells. All studies performed to date find that PAX3-FOXO1 is constitutively nuclear, even in the presence of activated AKT. However, whereas one transfection study did not find any changes in PAX3-FOXO1 transcriptional activity in the presence of activated AKT, a more recent study of a murine model of ARMS found that AKT hyperactivation results in PAX3-FOXO1 phosphorylation and decreased transcriptional activity. Studies in human ARMS cells are needed to resolve these issues and discern whether AKT activation has any effect on PAX3-FOXO1 function.
Modulation of Growth and Apoptotic Pathways by Fusion Oncoproteins
Gene transfer experiments have investigated specific functions of the PAX3-FOXO1 and PAX7-FOXO1 fusion proteins and suggest that these proteins exert an oncogenic effect through multiple pathways. In initial studies of growth control, transduction of PAX3-FOXO1 into murine NIH 3T3 or chicken embryo fibroblasts resulted in transforming activity, whereas wild-type Pax3 did not transform these cells. In a complementary study, a protein consisting of the N-terminal region of PAX3 fused to the KRAB transcriptional repression domain reverted the transforming activity of ARMS cells in culture and suppressed tumor formation of ARMS cells in mice. In mutagenesis analyses, the homeodomain but not the paired box is needed by PAX3-FOXO1 for transformation; thus target genes with paired box binding sites may not be required for cellular transformation. In addition, transforming activity is activated when the VP16 activation domain is substituted for the C-terminal domain of PAX3, suggesting that other activation domains can mimic the effect of the C-terminal FOXO1 domain.
More recent studies indicate that there is an antagonistic balance between transforming activity and growth-suppressive or toxic activity in many cell types in which PAX3-FOXO1 is expressed. Transforming activity is optimally exerted at low expression levels of exogenous fusion protein. In contrast, at higher expression levels, comparable to the levels in human ARMS tumors, PAX3-FOXO1 causes cell death or growth suppression in various nontransformed cell lines. Therefore human ARMS cells can tolerate these high “physiologic” expression levels, whereas the non-ARMS cells do not tolerate these higher levels. The hypothesis proposed is that additional genetic alterations are necessary in ARMS to attenuate the toxic and growth-suppressive effects of the fusion protein. One such event may be inactivation of the CDKN2A tumor suppressor gene that collaborates with PAX3-FOXO1 to permit primary human skeletal muscle cell precursors to bypass the senescent growth arrest checkpoint. It is noteworthy that mutation studies show that the growth suppressive and toxic activity is at least partly dependent on an intact paired box but does not require an intact homeodomain. These findings suggest that there may be two separate sets of target genes mediating the transforming and toxic phenotypes as a result of the complex function of the PAX3-FOXO1 DNA-binding domain.
Despite the ability of PAX3-FOXO1 to induce toxic effects when introduced into many cultured cells, an important function of the PAX3-FOXO1 oncoprotein in ARMS cells is maintenance of cell viability by inhibiting apoptosis. Treatment of ARMS cells with either an antisense oligonucleotide directed against the PAX3 translational start site or with siRNAs directed against the 5′ PAX3 region resulted in a decrease in PAX3-FOXO1 protein expression. This expression change was associated with a significant decrease in cell number as well as with morphologic and biochemical characteristics of apoptosis. Similarly, expression of a tamoxifen-inducible PAX3-KRAB construct in ARMS cells demonstrated evidence of apoptosis when induced in low serum conditions or tumor xenografts. One downstream transcriptional target of PAX3-FOXO1 that at least partially mediates this apoptotic function is the gene that encodes the transcription factor TFAP2B. The functional role of TFAP2B is indicated by the induction of apoptosis in ARMS cells when this gene is downregulated by siRNA and by the prevention of apoptosis mediated by siRNA directed against the 5′ PAX3 region when a construct constitutively expressing TFAP2B is introduced into ARMS cells.
Metastatic Pathways in Rhabdomyosarcoma
Early studies of RMS metastasis examined subclones of the RD ERMS cell line. The metastatic capacity of different subclones did not correlate with the tumorigenic or proliferative capability of the subclones. After exposure to conditions that stimulate myogenic differentiation, some subclones showed increased differentiation manifested by an increased fraction of myosin-positive cells. Although proliferative capability did not correlate with this in vitro differentiation, the metastatic efficiency was reduced in subclones demonstrating differentiation.
Additional studies with the RD line showed that its metastatic efficiency is affected by alterations of cell surface molecules. In the case of the cell surface glycoprotein NCAM (neural cell adhesion molecule), this protein is posttranslationally modified by the addition of polysialic acid on its extracellular domain. When the enzyme endoneuraminidase-N was injected intraperitoneally to cleave the polysialic acid from the surface of intraperitoneally implanted RD cells, there was a decrease in the frequency of lung and liver metastases. When the integrin VLA-2 (α2β1) that is not normally expressed by RD cells was introduced into these cells, there was increased adhesion to collagen and laminin in vitro and more metastatic foci in mice after either intravenous or subcutaneous injection.
Among the various RMS subtypes, there are differences in matrix metalloproteinase (MMP) expression that may contribute to differences in metastatic behavior. Immunohistochemical analysis of 5 MMPs in 33 RMS cases found higher expression of MMP1, MMP2, and MMP9 in ARMS than in ERMS cases. After confirming the higher MMP2 expression in ARMS in a comparison of ARMS and ERMS cell lines, the high MMP2 expression was correlated with more invasive behavior in the ARMS cell lines. In a human RMS cell line with spontaneous metastatic progression, there was upregulation of MMPs and downregulation of tissue inhibitors of metalloproteinases. Finally, attachment of RMS cells to fibronectin results in increased MMP2 expression and in vitro invasive behavior along with increases in COX-2 expression and prostaglandin E 2 production. Treatment with exogenous prostaglandin E 2 can recapitulate this effect and increase MMP2 expression at the level of the MMP2 promoter. In contrast, treatment of cells with COX-2 inhibitors can reverse the fibronectin effects on MMP expression and invasive behavior. Another soluble factor released by RMS cells is heparanase, which is found upregulated in several human RMS cell lines. Heparanase cleaves heparan sulfates, thus releasing and likely making more bioactive heparan-bound growth factors. shRNA-mediated suppression of heparanase in RD and Rh30 cells (a commonly studied human ARMS cell line) inhibited their invasive properties as assessed by Matrigel invasion assays. Parallel studies examining heparanase mRNA expression in human tumor samples and human serum in order to validate the upregulation were suggestive but not statistically significant because total numbers were small and error bars wide, thus suggesting that further studies will be required.
Three cell surface receptors, MET, CXCR4, and LIFR, are expressed in RMS and function in signaling pathways that impact metastatic behavior ( Fig. 59-6 ). Although MET and CXCR4 genes are downstream targets of the PAX3-FOXO1 fusion protein, all three genes are expressed in both ARMS and ERMS tumors. CXCR4 is a G-protein–coupled chemokine receptor whose ligand is stromal derived factor-1 (SDF1), whereas MET is a member of the tyrosine kinase family of receptors whose ligand is hepatocyte growth factor/scatter factor (HGF/SF). Finally, leukemia inhibitory factor (LIF) binds to a heterodimeric membrane receptor composed of the LIF-specific LIFR and the gp130 receptor chain, which is also used as the receptor for interleukin (IL)-6, oncostatin M, and several other cytokines. All three ligands are secreted by the bone marrow, an important site of ARMS metastasis. The CXCR4-SDF1 signaling pathway is involved in the homing of normal cells to hematopoietic sites, and the MET-HGF/SF signaling pathway is involved in the proliferation and motility of various cell types. The LIF-LIFR signaling pathway has a variety of roles in multiple tissues, including proliferation of hematopoietic cells and muscle satellite cell production. For these reasons, usurpation of these signaling pathways has been proposed in the metastasis of various cancers.
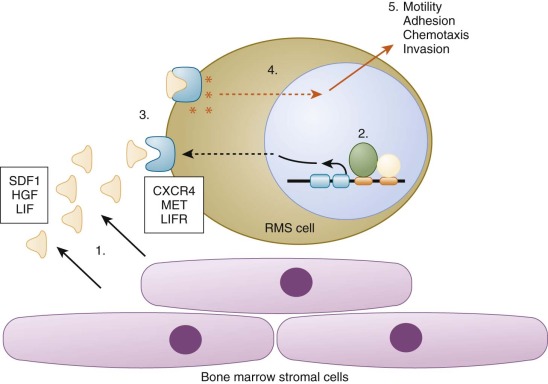
Cell culture studies have explored the influence of CXCR4-SDF1, MET-HGF/SF, and LIFR-LIF signaling on the metastatic behavior of ARMS cells. SDF1, HGF, or LIF treatment of RMS cell lines induced cell culture changes in relevant properties, including motility, adhesion, chemotaxis, and invasion. In an in vivo experiment, more ARMS cells than ERMS cells are chemoattracted to and seed lethally irradiated bone marrow in association with the upregulation of HGF and SDF1 in irradiated bone marrow stroma. In addition, a selected ARMS subclone that preferentially responds to LIF demonstrates increased seeding of bone marrow, liver, and lymph nodes after intravenous injection and increased cells in bone marrow and lung 6 weeks after intramuscular injection. Recently, another receptor for SDF-1 called CXCR7 has been identified. This receptor binds not only SDF-1 but also interferon-inducible T-cell α chemoattractant (ITAC) chemokine. It is expressed in both ARMS and ERMS cells and appears to be independent from CXCR4, suggesting that CXCR4-SDF-1 axis targeting alone may not be enough. Finally, as models of pathway-specific potential therapeutic strategies, a CXCR4-specific inhibitor blocked SDF1-directed adhesion and chemotaxis in ARMS cells, an siRNA directed against LIFR inhibited in vivo metastasis of ARMS cells, and suppression of MET receptor by shRNA in RMS cells decreased the migratory response in vitro.
Recently, three additional cell surface receptors including IL-4 receptor-α (IL-4Rα), FGFR4 (discussed in “ Oncogene and Tumor Suppressor Gene Mutations in Rhabdomyosarcoma ”), and cannabinoid receptor 1 (CNR1) have been implicated in RMS metastasis. IL-4Rα is found upregulated in human and murine ARMS, and blockade by a neutralizing antibody inhibited both lymphatic and hematogenous metastasis in a genetically engineered murine model of ARMS. Because there are neutralizing antibodies to IL-4R (and cognate ligands IL-4 and IL-13) in clinical use for nononcologic human diseases, this may be a feasible approach toward blocking RMS metastasis. FGFR4 gain of function (K535 and E550 tyrosine kinase domain) mutants enhanced the metastatic phenotype when expressed in both murine RMS cell lines and NIH 3T3 cells, as assessed by lung metastasis burden in xenograft assays. Lastly, CNR1, which is also a PAX3-FOXO1 target, was shown to be dispensable for PAX3-FOXO1–driven cell proliferation and transformation but required for invasion and metastasis. In transformed primary mouse myoblasts expressing PAX3-FOXO1, either genetic or pharmacologic suppression of CNR1 reduced cell invasion in vitro and lung metastasis in vivo.
Studies of a murine metastatic RMS model system elucidated an important pathway involving the Vil2 (encoding Ezrin) and Six1 genes. In this model system, RMS tumors developed in transgenic mice that were deficient in CDKN2A (Ink4/Arf) and overexpressed HGF/SF. In a comparison of gene expression profiles of highly and poorly metastatic cell lines derived from this system, 44 differentially expressed genes were identified (28 overexpressed and 16 underexpressed in highly metastatic cells), including the overexpressed genes Vil2 and Six1. Ezrin is an adhesion molecule and member of the ERM family that promotes cytoskeletal reorganization in pathways linked to survival, motility, invasion, and adherence. Six1 is a homeodomain-containing transcription factor involved in the development of several lineages, is a downstream target of PAX3-FOXO1 in NIH 3T3 cells, and is expressed by ARMS cells. Furthermore, the gene encoding Ezrin is a direct transcriptional target of Six1. These gene expression relationships were confirmed by RMS tumor studies in which both genes were generally expressed at higher levels in fusion-positive RMS tumors than in fusion-negative RMS tumors.
Expression of Vil2 and Six1 in RMS tumors was correlated with clinical stage, which is consistent with a role in tumor progression and metastatic behavior. There is direct proof of this role: transfer of either gene into low metastatic RMS lines from the HGF transgenic system caused increased metastatic activity. Similarly, when expression of either protein was suppressed by shRNA or repressed by a dominant-negative inhibitor, metastatic activity in the highly metastatic lines decreased. Finally, Ezrin is needed for Six1 to exert its metastatic function, thus further confirming that Ezrin acts downstream of Six1. These studies provide the framework of a pathway that leads from the fusion proteins to Six1 to Ezrin to metastatic activity.
There are surely additional genes and proteins supporting the metastatic phenotype in RMS, and to this end a European collaborative group evaluated in RMS the expression of genes known to support metastasis using an Affymetrix approach. After examining 19 primary human RMS tumor samples, they focused on two genes known as LMO4 and FOFOXF1. Although they did not evaluate for expression of the protein products of these genes in RMS samples, they did assess their role in metastasis by showing that siRNA suppression of these genes in RD and Rh30 cells decreased migration as assessed by Matrigel invasion.
Preclinical Investigation of Emerging Drug Targets for Rhabdomyosarcoma
Preclinical Models to Study Rhabdomyosarcoma Tumorigenesis
Human Tumor Cell Xenografts in Immunodeficient Mice
Although the identification of mutations and/or dysregulated signaling in RMS is accelerating, the value of these entities as therapeutic targets must be evaluated in preclinical models before application in humans. The original models to study cancer treatments other than in cell lines were xenograft models, in which human cell lines were injected as xenografts into immunocompromised mice. Historically for RMS this began at St. Jude’s Research Children’s Hospital in the 1980s and has evolved into the Pediatric Preclinical Testing Program ( http://pptp.nchresearch.org/ ). This program is a large-scale effort to systematically evaluate new agents against a panel of human RMS and other pediatric cancer cell lines by testing novel drugs against not only classic oncogene and tumor suppressor pathways including TP53 and receptor tyrosine kinases (RTKs) but also against newly identified targets found dysregulated in childhood cancer. Recently, the feasibility of adding radiation therapy to the Pediatric Preclinical Testing Program was demonstrated, using RMS as a pilot.
There are obvious advantages to examining the efficacy of novel agents against pediatric tumor cell lines, including that they are of human origin and have been validated in prior biologic studies. But there are disadvantages as well, including that many cell lines have been passaged in culture for decades, acquiring genetic changes not representative of the original tumor. There is also the practical problem of cell line annotation ; however, with the development of methods to fingerprint cell lines, identification has been more reliable. Finally, the intrinsic property that enables evaluation of human cell lines in a whole murine organism— their immunodeficient state—does not permit the evaluation of the role of the immune system in tumorigenesis, a problem that can be overcome by the use of genetically engineered mouse models.
Genetically Engineered Murine Models of RMS
With the advent of genetic manipulation of the murine genome, it is now possible to examine the genesis and pathophysiology of RMS at the organism level. Indeed there are more than two dozen genetically engineered murine models that give rise to RMS. The earliest models were serendipitously discovered, for example that ERMS occurs in TP53 knockout mice or SV40 transgenic mice. Later models were deliberate in their design and incorporated genetic lesions found in RMS, including overexpression of the cMET signaling pathway or PAX3-FOXO1 fusion gene. These and others are reviewed elsewhere. More recently, sophisticated models have emerged in which Cre-Lox (or Flp-Frt) recombinase technology has been harnessed to turn on or turn off expression of specific genes or negative regulatory components such as shRNAs. This has been especially useful in examining the cells of origin of RMS, which may range from an undifferentiated mesenchymal stem cell to the myonucleus of a differentiated myofiber. These approaches have led to some surprising discoveries, such as the finding that RMS can arise from the adipocyte lineage and that ERMS is on a continuum with a common adult sarcoma known as undifferentiated pleomorphic sarcoma. Last, these genetically engineered mouse models have also been useful as additional models for preclinical studies, such as the PAX3-FOXO1– based ARMS model.
Lower Eukaryote Models of RMS
Although murine models have been the mainstay of modeling human cancer including RMS, lower organisms including zebrafish (Danio rerio) and the fruitfly (Drosophila melanogaster) are more amenable to genetic manipulation and screening and contribute a unique understanding of RMS genesis and metastasis. For example, zebrafish models of ERMS have been useful for in vivo imaging of tumor-propagating cells and their properties and understanding the impact of oncogenic RAS expression at different stages of muscle development. Drosophila models of ARMS have provided insight into the pathophysiology of PAX-FOXO1 fusion genes and the role of defective myoblast fusion in RMS.
A comprehensive listing of RMS animal models and their utility is available. Along with the xenograft and genetically engineered mouse models, these models have enabled testing of pharmacologic agents in a preclinical fashion before to moving to phase I clinical trials. Below are listed some examples of agents that have been tested in a variety of in vivo preclinical models.
Inhibitors of Receptor Tyrosine Kinases
RTKs are embedded in the cellular plasma membrane and transmit signals from the environment to the cell interior. When a ligand binds to an RTK, dimerization occurs and this leads to internal phosphorylation events and signaling through a variety of canonical intracellular signaling pathways such as AKT, MAPK, mammalian target of rapamycin (mTOR), and many others. Because RTKs are at the cell surface, they can be blocked with interfering antibodies (or their ligands can be inhibited with neutralizing antibodies). Given the expansion in our understanding of the RTKs that are dysregulated or upregulated in RMS, there are several monoclonal antibodies that have been evaluated in RMS. The prototype was a monoclonal targeting IGFR-1, known as figitumumab (previously CP-751871). This agent not only blocked binding of ligand to the IGFR-1 but also downregulated the receptor, suppressed signaling to AKT, inhibited tumor-derived VEGF, and synergized with rapamycin. Several other studies followed, including testing of other anti–IGFR-1 monoclonal antibodies, some of which advanced to clinical trials. However, tumor cells were able to activate bypass routes, rendering resistance to IGFR-1 blockade. Bevacizumab, a monoclonal antibody to VEGF, has been evaluated in a phase II clinical trial for RMS (see “ Clinical Research in Rhabdomyosarcoma ”). Still in development and preclinical testing are monoclonal antibodies to FGFR4 and NCAM, both upregulated in a significant percentage of RMS tumors and implicated in RMS metastasis (see “ Metastatic Pathways in Rhabdomyosarcoma ”). Other new agents are also being considered for RMS.
Given their enzymatic activity, RTKs can also be inhibited with small molecules. In a manner similar to the prototype small molecule imatinib, which was developed to inhibit the BCR-ABL fusion protein in chronic myelogenous leukemia, there are also small molecule inhibitors being evaluated for RMS-associated RTKs. An early example was BMS-754807, an oral small molecule inhibitor of IGFR-1, which showed intermediate efficacy in RMS. Later examples include sunitinib, cediranib, and pazopanib, which are small molecules that inhibit the kinase activities of platelet-derived growth factor receptor (PDGFR) and other RTKs, VEGF receptors 1 to 3 (VEGFR1 to VEGFR3), and VEGFR1 to VEGFR3 and other RTKs, respectively. Given the known resistance that emerges with RTK monotherapy, these small molecules are now being tested in conjunction with traditional chemotherapeutic agents. For example, pazopanib has been tested in combination with topotecan, and in Rh30 xenografts this combination inhibits tumorigenesis. Indeed, this has supported the incorporation of pazopanib into human clinical trials (see “ Clinical Research in Rhabdomyosarcoma ”). Whereas the upregulation of a specific RTK in RMS warrants investigation of that RTK as a drug target, not all RTK inhibitors have efficacy. For example, although EGFR is found upregulated in some RMS tumors, erlotinib (an EGFR inhibitor) had no effect on blocking tumorigenesis in a genetically engineered mouse model of ARMS.
In addition to inhibiting RTKs, whether through monoclonal antibodies or small molecules, there are also efforts to inhibit heparanases (discussed in “ Metastatic Pathways in Rhabdomyosarcoma ”). Pharmacologic inhibition of heparanases using SST0001, a chemically modified heparin, in RD and Rh30 xenografts showed inhibition of tumor growth. The precise mechanism is not clear, but there was evidence of decreased angiogenic factors secreted from the RMS cell lines in the in vitro studies.
Inhibitors of Intracellular Signaling Pathways
Regarding downstream signaling pathways, it is clear that blockade of only one pathway is not durably antitumorigenic because of compensatory upregulation of other pathways. Therefore there are ongoing efforts to simultaneous block two or more pathways. Dual blockade of the PI3K/AKT/mTOR and RAS/MEK/ERK pathways with small molecules synergistically inhibits RMS cell growth in vivo. Similarly, treatment of RMS xenografts with rapamycin was shown to block tumor growth by inhibiting not only the mTOR pathway but also Hedgehog signaling, and indeed rapamycin analogues are in human clinical trials (see “ Clinical Research in Rhabdomyosarcoma ”). Attesting to the power of utilizing lower eukaryotes for target discovery, a screen performed in an oncogenic RAS-driven model of zebrafish RMS identified dual MEK and mTOR/S6K1 inhibition as effective.
Finally, there is RAS itself, which is found mutated in a significant portion of ERMS tumors (see previous section, “ Oncogene and Tumor Suppressor Gene Mutations in Rhabdomyosarcoma ”). Targeting the RAS protein has proven difficult. However, silencing of Sprouty1, an upstream modulator of RAS that is also found in ERMS tumors and associated with upregulated ERK signaling, using shRNA triggers complete regression of ERMS xenograft tumors carrying a mutated oncogenic RAS gene, suggesting that there will be alternate ways to block RAS signaling in the future.
Inhibitors of Developmental Pathways
Pharmacologic blockade of RTK signaling by small molecule inhibitors became possible because of a deep structural understanding of their enzymatic activity. As the role of the evolutionarily conserved developmental pathways (e.g., Hedgehog, Notch, Wnt, transforming growth factor-β [TGF-β]) in RMS tumorigenesis has become apparent, it has become similarly imperative to understand the molecular basis for their dysregulation. For the most part, it appears that in RMS these pathways, which during normal development and tissue homeostasis direct myogenesis by promoting myogenic stem cell maintenance, expansion, and tight control of differentiation, are usurped and prevent proper myogenic lineage progression. In this regard, agents developed to inhibit members of these pathways must be evaluated for unanticipated effects on normal tissue, especially considering the continual growth and development of pediatric patients. Studies in Ptch mutant mice, which through increased Hedgehog signaling develop both medulloblastoma and RMS, showed that combined dosing with the epigenetic drugs 5-aza-2′-deoxycytidine and valproic acid inhibited tumor formation when treated early. In human RMS xenografts, treatment with betulinic acid (a naturally occurring plant product), GANT61 (a Gli antagonist), or forskolin (also derived from a naturally occurring plant product) suppressed Hedgehog signaling and RMS tumor growth. Recently, several additional small molecule inhibitors of Smoothened and Gli have been developed, raising the expectation that blockade of Hedgehog signaling will be possible in human cancers.
Regarding Notch signaling, given its critical role in normal muscle development and the upregulation of many components of this pathway in RMS, this pathway has gained attention, and several Notch pathway genes and proteins have been suppressed using genetic (shRNA) or pharmacologic (γ-secretase inhibitors) approaches and when suppressed shown to inhibit the growth of RMS xenografts, including Notch1, Notch3, RBPJ, and Hey1. Although as important in skeletal muscle development as Notch, the Wnt pathway is not as well understood in the pathobiology of RMS and therefore in vivo preclinical studies using genetic or pharmacologic approaches to target the Wnt pathway are lagging. Finally, regarding TGF-β signaling, this pathway, too, is under investigation in RMS. However, recently a miR that is suppressed by TGFB1 known as miR 450b-5p, when overexpressed in RMS cells inhibits cell growth in vitro and tumor xenografts in vivo and promotes myogenic differentiation.
Inhibitors of Other Cellular Processes That Support Rhabdomyosarcoma Tumorigenesis
Apoptosis
In addition to specific signal transduction pathways, there are efforts to target specific cellular processes. For example, as described earlier, RMS cells can resist apoptosis (programmed cell death) through upregulation of anti-apoptotic proteins or the suppression of pro-apoptotic proteins. Therapeutic approaches therefore may include inhibiting anti-apoptotic signaling, for example through neutralizing Bcl-2 family proteins (although caution must be taken because some Bcl-2 family members have pro-survival roles), or activating pro-apoptotic signaling, for example through stimulation of the extrinsic apoptotic pathway via members of the tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptors. Drozitumab is a monoclonal antibody to death receptor 5 that supports assembly of death-induced signaling complexes and has shown efficacy in RMS xenografts. It may also be possible to reestablish normal function of mutated pro-apoptotic pathways. For example, in ARMS xenografts the MDM2 inhibitor RG7112 interferes with MDM2-TP53 binding, releasing TP53 from negative control and permitting its normal cell surveillance function. Finally, although apoptosis has historically been the major focus of inducing cell death, the process of necroptosis (nonapoptotic cell death) has been found to be induced in RMS cells via obatoclax, which inhibits BCL2.
Although involved not only in avoidance of apoptosis but also many other pro-tumorigenic processes, it is important to note that MYCN has been proven targetable through the use of antigene therapy in which a peptide nucleic acid oligonucleotide (tethered to a nuclear localization signal peptide and targeted against the antisense strand of MYCN exon 2) was delivered to ARMS cells. This antigene therapy caused a decrease in proliferation and an increase in apoptosis in vitro and eliminated 75% of tumors in Rh30 RMS xenografts, with 25% showing a reduction in tumor.
Cell Cycle
Although apoptosis is a cellular event that can be triggered by many distinct stimuli, direct manipulation of the cell cycle and its checkpoints also provides opportunities for pharmacologic manipulation and treatment of RMS. Of long-standing interest are antimitotic agents, which interfere with transit through mitosis, including spindle assembly, chromosome positioning, and chromosome separation. Classic chemotherapy agents such as vincristine, which is a backbone of RMS therapy, interfere with the mitotic spindle by binding tubulin. Indeed, a more potent synthetic antitubulin agent, eribulin, which is distinct from the Vinca alkaloids in how it binds tubulin, has shown some complete responses in RMS xenografts. More recently, kinases and kinesin motor proteins that control mitosis are now able to be pharmacologically targeted and agents blocking these proteins are being evaluated against human cancer panels, including RMS. The agents MLN8237 and B16727, Aurora A, and Pololike kinase inhibitors, respectively, have both demonstrated some response in RMS xenograft studies. The kinesin spindle protein inhibitor ispinesib was toxic and ineffective in inhibiting RMS xenograft growth, whereas the CENP-E kinesin inhibitor GSK923295A showed some efficacy.
Regulation of Protein Stability and Turnover
The heat shock and ubiquitin-proteasome systems, which contribute to homeostasis of protein stability and turnover, have also been mined for their potential as anticancer targets. 17-DMAG, a first-in-class derivative of geldanamycin that inhibits Hsp90, showed some antitumor activity in xenograft models of ARMS, whereas a non-geldanamycin Hsp90 inhibitor, AT13387, showed no objective tumor responses. Bortezomib, which is approved by the U.S. Food and Drug Administration (FDA) for the treatment of several human diseases, showed activity in ERMS and ARMS xenografts in vivo. MLN4924, a first-in-class NEDD8-activating enzyme inhibitor that affects the ubiquitin-proteosome pathway also showed efficacy in RMS.
Epigenetic Modifiers
Currently there are two groups of agents under study that qualify as epigenetic modifiers. The first group of agents is composed of histone deacetylase (HDAC) inhibitors. Agents in the second group inhibit DNA methylation and include 5-aza-2′-deoxycytidine. Although results of early in vitro studies were promising, there are few in vivo studies demonstrating inhibition of RMS tumorigenesis in response to epigenetic modifiers. As mentioned earlier, combined dosing with the epigenetic drugs 5-aza-2′-deoxycytidine and valproic acid, which target Dnmt and HDAC, respectively, in a genetically engineered mouse model of Ptch- mutant mice inhibited tumor formation, but only when the mice were treated early during tumorigenesis. Additionally, drugs that target proteins involved in epigenetic regulation can have unanticipated protumorigenic effects, as in the case of the HDAC inhibitor trichostatin A and DNA demethylating agent (5-Aza), which reactivated the expression of Ezrin, a known prometastatic protein in RMS.
Immunotherapy for Rhabdomyosarcoma
In addition to pharmacologic agents, there are efforts to stimulate or harness the immune system to target and kill RMS tumor cells. However, like most other cancers, RMS is remarkably successful at evading the immune system. One approach to overcome this evasion is isolation and expansion of natural killer cells obtained from normal healthy donors, which can kill human sarcoma cells in vitro and Ewing sarcoma xenografts in vivo. Recently, IL-15 cytokine–induced killer cells from donors were shown to be active in xenograft models of RMS. Another novel approach is chimeric antigen receptor therapy, which although still in early phases has been effective in the treatment of leukemias. Thus there is hope that this approach can be similarly used in solid tumors. To prepare for these efforts, a recent study identified candidate cell surface proteins on pediatric tumors including RMS that could be future therapeutic targets for immunotherapy. Finally, there are ongoing efforts to develop vaccines to solid tumors including RMS. Given the specific expression of the PAX3-FOXO1 fusion protein in ARMS, an epitope-enhanced peptide bearing the PAX3-FOXO1 breakpoint was designed and generated. Because this peptide represents a neoantigen exclusively found in ARMS, the immune system should recognize it as such; and, indeed, dendritic cells pulsed with this peptide generated a CTL line that lysed human ARMS tumor cells in vitro. Similar proposals have been made to use the fetal acetylcholine receptor and other antigens for RMS immunotherapy.
Related posts:
 The Neonatal Erythrocyte and Its Disorders
The Neonatal Erythrocyte and Its Disorders
 Diagnostic Approach to the Anemic Patient
Diagnostic Approach to the Anemic Patient
 Approach to the Child with a Suspected Bleeding Disorder
Approach to the Child with a Suspected Bleeding Disorder
 Disorders of the Red Cell Membrane
Disorders of the Red Cell Membrane
 Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
 Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


