products of the MHC on the surface of antigen-presenting cells (APCs). Soon after the existence of T cell-mediated suppression was appreciated, some studies suggested that interactions among multiple distinct T-cell subpopulations might be involved. A CD4+ cell was described that appeared to induce CD8+ suppressor cells and was called the suppressor inducer cell. Contrasuppressor T cells had no independent helper, suppressor, or cytotoxic activity on an immune response, but enhanced immune responses by preventing the downregulation mediated by suppressor cells.
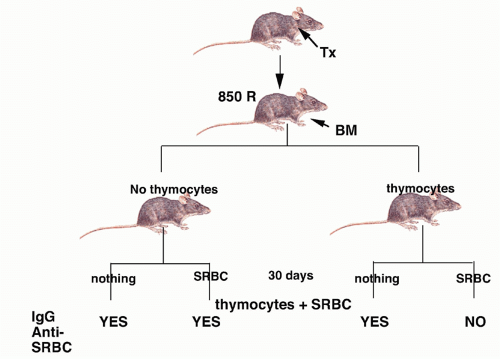 FIG. 33.1. First Demonstration of Suppressor T Cells. Adapted from Gershon and Kondo.3 |
T cell factors. These studies, together with the inability to identify a marker specific for suppressor T cells and the inability to purify suppressor T cells, raised considerable doubts about the existence of a distinct functional lineage of suppressor T cells.
 |
the helper population responsible for autoantibody production intact. The disease model was autoimmune thyroiditis because circulating antibody to thyroglobulin was believed to play an important pathogenic role. Spontaneous thyroiditis and circulating IgG autoantibodies developed in 60% of rats following the selective depletion of T cells by adult Tx followed by irradiation. Tx was performed between 3 and 5 weeks of age, and the rats were then given four to five repeated doses of 200 rad at 14-day intervals (Fig. 33.3). No evidence of thyroiditis was seen in rats that received only local irradiation to the thyroid region, indicating that irradiation itself did not induce pathologic changes. The conclusion drawn from these studies was that in the normal animal, B cells that recognized thyroid antigens were prohibited from differentiating into autoantibody-producing cells by an active controlling T cell mechanism. It was assumed that the suppressor T-cell population was mediating its functions by acting directly on the B cell and not by regulating other T cells. The active role of T cells in preventing the development of autoimmunity in this model was confirmed by reconstituting, shortly after the final dose of irradiation, the Tx-irradiated mice with lymphoid cells from normal donors. Penhale and colleagues19 also demonstrated that autoimmune diabetes would develop following the Txirradiation protocol in a strain of rats that was normally not susceptible to this disease. Taken together, the d3Tx model in the mouse and the Tx-irradiation model in the rat demonstrated that normally autoreactive helper and suppressor cells may coexist and that certain autoimmune responses are held in check by the equilibrium favoring suppressor activity.
TABLE 33.1 Organ-Specific Autoimmune Disease and Lymphopenia | ||
|---|---|---|
|
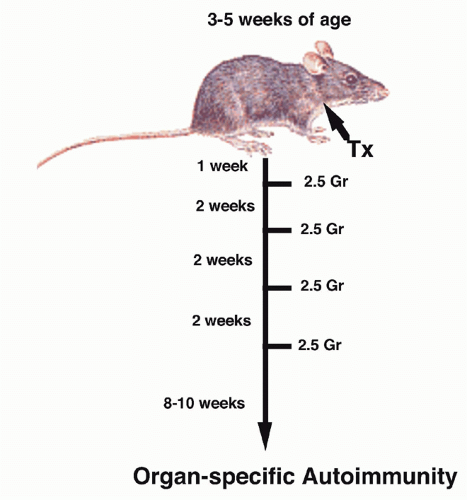 FIG. 33.3. Induction of Organ-Specific Autoimmunity in Rats by Adult Thymectomy and Irradiation. Adapted from Penhale et al.17 |
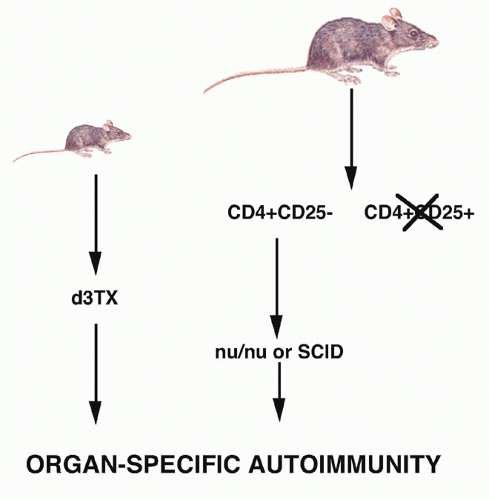
the role of Treg cells was the demonstration by Sakaguchi et al.,21,22 that a minor population of CD4+ T cells (10%) that coexpressed the CD25 antigen (the interleukin [IL]-2R α-chain) appeared to function as Treg cells in the normal adult. When CD25+ T cells were depleted from a population of normal adult CD4+ T cells and the remaining CD4+CD25- T cells transferred to an immunocompromised recipient such as a nu/nu mouse, the recipients developed a spectrum of autoimmune diseases that closely resembled those seen following d3Tx (see Fig. 33.2). Cotransfer of the CD25+ cells prevented the development of autoimmunity. Similarly, the induction of disease post-d3Tx could also be prevented by reconstitution of the animals with CD4+CD25+, but not CD4+CD25-, normal adult T cells before day 14 of life.23 CD8+CD25-T cells alone were not capable of inducing autoimmunity and enhanced the induction of disease induced by CD4+CD25- T cells. These studies solidified the role of the CD4+CD25+ T cells as a major subset of cells that plays a unique role in the regulation of the immune response. The autoimmune diseases induced by depletion of CD4+CD25+ T cells are uniformly accompanied by the development of organ-specific autoantibodies, suggesting that this mode of loss of T-cell tolerance also results in the breakdown of B-cell tolerance as well. It is likely that the activated self-reactive T helper cells provide signals to self-reactive B cells, rescue them from apoptosis, and stimulate autoantibody production.
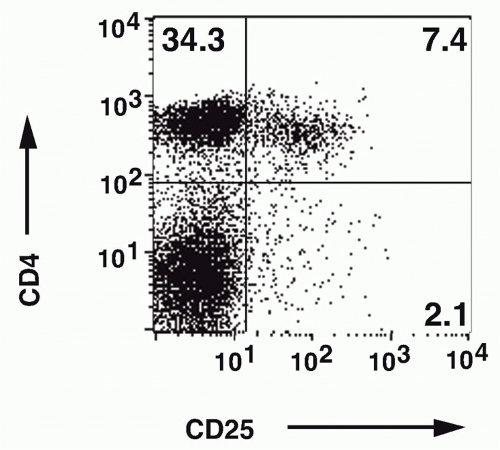 FIG. 33.4. Identification of CD4+ CD25+ T Cells in Normal Mouse Lymph Node. Adapted from Thornton and Shevach.37 |
Foxp3, was identified as the key player in the biology of CD4+CD25+ Tregs.42,43,44,45 Young males with the immune dysregulation polyendocrinopathy enteropathy X-(IPEX) linked syndrome or a mutant mouse strain, scurfy, succumb to similar autoimmune and inflammatory diseases as a result of uncontrolled activation and expansion of CD4+ T cells. Both IPEX and scurfy mice have mutations in a common gene, Foxp3, which encodes a forkheadwinged-helix transcription factor. There is an excellent correlation between expression of Foxp3 and CD25, but a minor population (˜10%) of Foxp3+ cells is CD25- and about 10% of CD25+ cells are Foxp3- (Fig. 33.5); the latter represent activated effector cells. Expression of Foxp3 only in the thymus using a proximal lck-driven transgene does not prevent disease in scurfy mice. Foxp3 expression in peripheral T cells is thus required for maintenance of Treg function. Retroviral-mediated transduction of Foxp3 expression in naïve T cells can convert these cells to a regulatory phenotype functioning in vitro in a manner similar to CD4+CD25+ Tregs. Furthermore, Foxp3-transduced naïve CD25- T cells also manifest suppressor activity in vivo and can inhibit the weight loss, diarrhea, and histologic development of colitis induced by transfer of CD25-CD45RBhigh cells as effectively as Tregs.
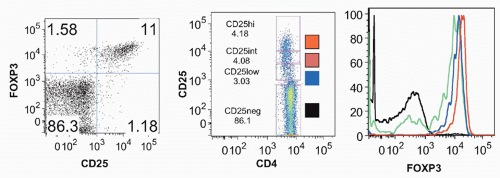 FIG. 33.5. Correlation between CD25 Expression and Foxp3 Expression in Mouse CD4+ Cells. |
decreased level of Foxp3 protein in Treg cells as a result of induced dysregulation of Foxp3 gene transcription results in marked attenuation of Treg suppressor function.49
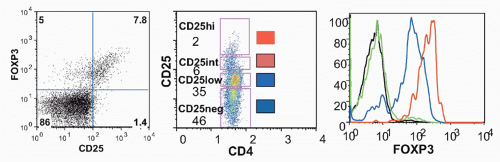 FIG. 33.6. Correlation between CD25 Expression and Foxp3 Expression in Human CD4+ Cells. |
large proportion expressed CD25 and functioned as Tregs.54 Radioresistant elements of the thymus were shown to be both necessary and sufficient for the selection of Foxp3+ T cells in these doubly transgenic mice. Similar results were obtained when TCR transgenic mice on a RAG-/- background were mated to the HA transgenic mice, clearly indicating that thymocytes that can only express a single transgenic TCR can undergo selection to become Tregs. A second TCR transgenic mouse was generated that expressed a variant determinant of HA, but that recognized the S1 determinant with 100-fold less affinity. When these mice were bred to the HA transgenic mouse (S1), they did not have an increased frequency of Foxp3+ T cells. Thus, thymocytes with a low intrinsic affinity for the S1 peptide did not develop into Foxp3+ thymocytes in response to HA. These data are consistent with a model in which selection of Tregs that express a transgenic TCR depends on a high-affinity interaction of the TCR with its ligand. Treg differentiation results from interactions that lie in between the signaling strength required for conventional positive selection on one side and clonal deletion on the other side. It could not be determined from these studies whether this selection process occurs on cortical or medullary epithelial cells.
be secondary to the capacity of Foxp3- T cells to produce IL-2,75 or to some intrinsic defect in the Foxp3+ T cells in their capacity to respond to IL-2. IL-2Rα-/- T cells in mixed bone marrow chimeras had a substantially decreased competitive fitness compared to WT Foxp3+ Tregs. The conclusion drawn from these studies was that IL-2 is an essential Treg cell growth factor in vivo and that in the absence of IL-2, Treg cells exhibit an impaired metabolic fitness. It appears that Foxp3 expression in Tregs establishes a gene expression program that renders Tregs critically dependent on paracrine IL-2 signalling resulting in repression of IL-2 production and the induction of IL-2Rα. IL-2 is not only required for the survival of Foxp3+ Tregs in the periphery, but also appears to be critical for maintenance of their suppressive functions.76,77,78 Deletion of the proapoptotic factor, Bim, results in the restoration of normal numbers of Foxp3+ T cells in IL-2-/- mice, but Bim-deleted mice still exhibited lethal autoimmunity.78
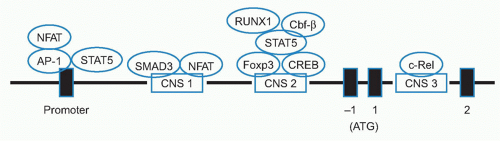 |
in detectable tissue specific autoimmunity in CNS1-/- mice until 1 year of age. These results suggest that mechanistic requirements for thymic and peripherally induced Tregs are distinct from Tregs differentiated in the thymus.
in the presence of TGF-β, both RORγT and Foxp3 are transcribed and interact with each other. In the presence of IL-2, Foxp3 expression dominates leading to suppression of RORγT transcription, promoting Treg development. In contrast, in the presence of IL-6, and low levels of IL-2 the transcription of RORγT is favored which in turn represses Foxp3 transcription with resultant Th17 devlopment.101
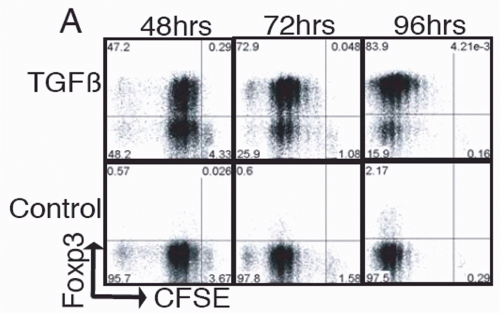 FIG. 33.8. Naïve Foxp3- T Cells were Cultured and Stimulated in the Presence of Transforming Growth Factor-χ. Foxp3 expression was determined by intracellular staining at the indicated times. Adapted from Davidson et al.100 |
tolerance.105,106 In addition to the TGF-β induction pathway, Foxp3+ Tregs can also be induced via PD-L1-PD-1 ligand receptor pathway107 and by environmental factors such as some aryl hydrocarbon receptor agonists.108
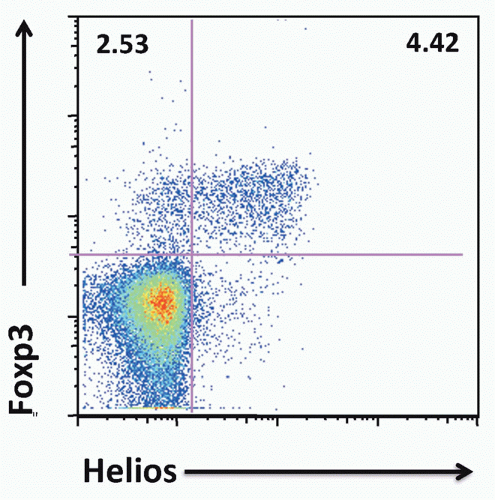 FIG. 33.9. Approximately 70% of Mouse Foxp3+ T Cells Express Helios. Adapted from Thornton et al.119 |
with their functional properties. Most of the results are consistent with the latter possibility. Some genes are differentially expressed between the resting Foxp3- T cells and Tregs, while others are differentially expressed following activation.
TABLE 33.2 Genes Selectively Activated in CD4+CD25+ T Cells | ||
|---|---|---|
|
TABLE 33.3 Transcription Factors Bound and Regulated by Foxp3 | |||||
|---|---|---|---|---|---|
|
to Foxo1 and Foxo3, Treg cells were selectively enriched in genomic fragments containing the CNS1 and CNS3 regions, but not the CNS2 region of the Foxp3 promoter. Foxo transcription factors constitute one element of higher order control132,133 of Treg development and function.
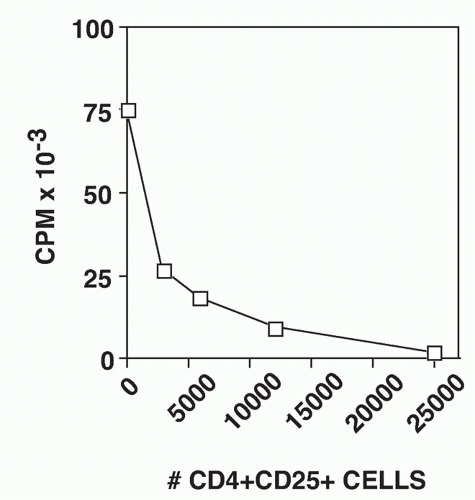 FIG. 33.10. CD4+ CD25+ T Cells Suppress the Proliferative Response of CD4+CD25-T cells. Graded numbers of CD4+CD25+ T cells were mixed with 5 × 104 CD4+CD25- T cells, T-depleted spleen cells, and soluble anti-CD3. Proliferation was measured after 72 hours. Adapted from Thornton and Shevach EM.37 |
culture in IL-2, activated Foxp3+ T cells remain nonresponsive and cannot be induced to proliferate when restimulated via their TCR in the absence of IL-2. The activated Foxp3+ T cells have more potent suppressor activity on a per cell basis (three- to fourfold) than freshly explanted Foxp3+ T cells. Foxp3+ T cells that appear to be antigen-specific can be identified in mice that express a transgenic TCR. In general, the percentage of Foxp3+ cells is reduced in TCR transgenic mice (3% to 5% compared to 10% in normal animals). The expression of the transgenic TCR α-chain is a convenient tool that allows activation of the Foxp3+ T cells with peptide/MHC rather than with anti-CD3.136 When Foxp3+ T cells from TCR-transgenic mice are activated with their peptide/MHC ligand and then expanded in vitro in IL-2, the activated suppressors are subsequently capable of suppressing the proliferative responses of fresh Foxp3- T cells from mice that express a different transgenic TCR. There is no MHC restriction in the interaction of the activated suppressors and the Foxp3- responders. Therefore, the suppressor effector function of activated Foxp3+ T cells in vitro is completely nonspecific (Table 33.4).
TABLE 33.4 The Suppressor Effector Function of Activated CD4+ CD25+ T Cells is Completely Nonspecific | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
activation. The diminished activity of PLC-γ1 in Tregs is likely to contribute, not only to the delay and reduced appearance of NFAT in the nucleus, but also to the attenuated activation of the PKC- and Ras-ERK-directed pathways as well. Other studies145 have suggested that the proximal promoter of the IL-2 gene fails to undergo chromatin remodeling and remains in a closed chromatin configuration.
pathogenic Th1 responses by downregulating STAT-1 expression in Treg cells.159
TABLE 33.5 CD4+ CD25+ Suppressor Function Occurs Independently of Transforming Growth Factor-β | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
identified GARP (LRRC32) as the cell surface receptor for latent TGF-β. Although the GARP/Latent TGF-β complex is also expressed by platelets, within the immune system, GARP expression is most often observed on activated Foxp3+ Tregs (Fig. 33.11). However, the function of the GARP/latent TGF-β complex on Tregs has remained elusive. Tregs that lacked the surface expression of LAP following treatment with TGF-β siRNA or the expression of GARP and LAP following treatment with GARP siRNA were somewhat less suppressive than Tregs treated with the control siRNA in the in vitro suppression assay. The roles for secreted or cell surface associated TGF-β as major mediators of the in vitro or in vivo suppressive functions or other functions of Tregs remains to be further explored.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


