Introduction
Hormones exert their actions by binding to specific receptor proteins, a process that induces conformational changes or compartmental redistribution of these proteins. The activated receptor is now capable of inducing positive (or negative) intracellular effects that ultimately are recognized as a physiologic response. The specificity of hormone action is determined by the affinity of hormones for different receptors, the cell-specific expression of the receptor, and the unique responses induced by ligand occupancy.
Since the early 2000s, our understanding of hormone action has advanced rapidly with the success of genomics and advanced molecular biologic techniques. This combined approach has led to the discovery and classification of an unexpectedly large number of receptors, some quite novel and others even unanticipated, that are members of large families of genetically conserved proteins. Moreover, our understanding of receptor action has been clarified by the identification and detailed characterization of postreceptor signaling proteins and signaling mechanisms. Four major receptor superfamilies have been identified that are distinguished by protein structure, cellular localization, and effector systems. These families include the G protein–coupled receptors (GPCRs), cytokine receptors, tyrosine kinase receptors (RTKs), and nuclear receptors ( Table 3.1 ). This chapter reviews major features of these important receptor families. Mutations influencing receptor function leading to endocrine disorders are also highlighted.
| Receptor Class | Hormone Receptors |
|---|---|
| G protein–coupled receptors | ACTH and other melanocortins, V2 vasopressin, LH, FSH, TSH, GnRH, TRH, GHRH, corticotropin-releasing factor, somatostatin, glucagon, oxytocin, gastric inhibitory peptide, type 1 PTH, free fatty acid, GPR54, orexin, ghrelin, melanin-concentrating, calcitonin, glucagon-like peptide-1, and calcium-sensing receptors |
| Type 1 cytokine receptors | Growth hormone, prolactin, and leptin receptors |
| Receptor tyrosine kinases | Insulin, IGF-1, and fibroblast growth factor receptors |
| Nuclear receptors | Thyroid hormone, vitamin D 3 , PPARγ, HNF4A, glucocorticoid, androgen, estrogen, mineralocorticoid, and DAX1 receptors |
Basic principles of receptor action
A molecule that binds to a receptor is called a ligand . When ligand binding leads to activation of signaling processes inside cells that ligand is called an agonist . The response of a receptor to its ligand is generally assessed by two characteristics of the ligand: potency and efficacy. Potency describes the concentration of ligand needed to cause the biological effect by binding to the receptor. A potent agonist activates the receptors at a low concentration, typically in the nanomolar range. Efficacy describes the maximal effect induced by a ligand. Potency is generally described by using the EC 50 , which is the concentration of ligand that induces a half-maximal effect. When a synthetic agonist exceeds the efficacy of the natural ligand it is called a super agonist . When receptor ligands (natural or synthetic) do not induce full activation, they are called partial agonists . An antagonist is a molecule that blocks the natural ligand from binding to its receptor. If it binds to the same site as the natural agonist it is called an orthosteric antagonist . If it binds to a different site than the natural agonist, it is called an allosteric antagonist . The potency of the antagonist is described by the IC 50 , which is the concentration that causes half-maximal inhibition. A receptor that has constitutive basal activity can be bound by a ligand that inhibits the receptor’s activity in the absence of the natural ligand. In this case the ligand is called an inverse agonist . A scale has been formulated to express the continuity in receptor ligand function—from –1 (representing a full inverse agonist), to 0 (representing a neutral antagonist), to + 1 (representing a full agonist).
Receptors have multiple possible conformations that are constantly changing. Receptors exist in some conformations more than others, based on the free energy of each conformation. Agonists act as modulators that stabilize a given conformation of a receptor by reducing the energy needed to enter that conformation. This conformation is associated with activation of downstream signaling. Partial agonists stabilize the receptor in conformations less efficient at activation of downstream signaling. Super agonists stabilize the receptor in conformations more efficient at activation of downstream signaling. Conformational changes in a receptor can also affect the number and type of signals that a receptor generates. In the classic paradigm, a GPCR was believed to function as a binary switch that could be activated by agonist binding or inhibited by antagonist blockade of agonist binding. We now know that GPCR signaling is more complex than a simple binary switch model (i.e., “on” and “off”), and that different ligands can bind the same GPCR and selectively activate one downstream pathway versus another. This ability of different ligands to induce different confirmations of a receptor that activate (or inhibit) selective downstream signals is called biased signaling . Ligands that bind to the native site of a receptor but produce different signaling events are termed orthostatic ligands . An example of this is provided by the follicle-stimulating hormone (FSH) receptor, a classic GPCR. Fully glycosylated FSH is more acidic and acts as a full agonist at FSH receptors, activating Gs-coupled signaling and generation of cyclic adenosine monophosphate (AMP). Partially glycosylated FSH is more basic and acts as a partial agonist or a biased ligand as it activates both Gs and Gi pathways, which compete. Deglycosylated FSH has no effect on signaling but binds to the receptor and so acts as a competitive antagonist. These differently glycosylated variants exist in the circulation and are a means for fine-tuning the signal from the pituitary to the gonads. In certain cases, a neutral ligand can bind to the receptor at a different site than the natural ligand’s binding site and by doing so, influence the conformation of the receptor such that the ligand’s efficacy is increased, decreased, or biased. These are called allosteric modulators . They do not activate or inhibit the receptors on their own. Similarly, there are allosteric synthetic ligands for the FSH receptor that demonstrate signaling bias ranging from full agonists to reverse agonists.
Receptors are generally found associated with other proteins whether at the cell surface or in the cytoplasm. These associated proteins influence receptor conformation. An example is receptor activity modifying proteins or RAMPs. Calcitonin receptor and calcitonin-like receptor are GPCRs that can bind to several ligands (calcitonin, adrenomedullin, amylin, and calcitonin gene-related protein). RAMP1, RAMP2, and RAMP3 associate with both the calcitonin receptor and calcitonin-like receptor, and depending on which RAMP is associated determines selectivity for one of the aforementioned ligands.
Conformational fluidity of receptors and interactions with multiple ligands and modulators leads to greater complexity, specificity, and fine-tuning, as well as overall efficiency.
G protein–coupled receptors
More than 1% of the genome of vertebrates encodes a large protein family of receptors that sense molecules outside the cell and activate signal transduction pathways and, ultimately, cellular responses. These receptor proteins are embedded in the plasma membrane and are coupled to intracellular signal generating systems by heterotrimeric G proteins (i.e., GPCRs). GPCRs are also known as seven-transmembrane domain receptors , 7TM receptors , heptahelical receptors , and serpentine receptors . They are called transmembrane receptors because they pass through the cell membrane, and they are called seven-transmembrane receptors because they have alpha helical regions that pass through the cell membrane 7 times. The human genome encodes roughly 950 GPCRs. GPCRs are involved in many diseases and are also the target of approximately 40% of all modern medicinal drugs. Approximately 150 of the GPCRs found in the human genome have unknown functions. Most GPCRs are odorant and pheromone receptors. Also important to note is that most hormones bind to GPCRs, and hence G protein–dependent signal transduction represents the most common mechanism for hormone action ( Table 3.2 ).
| Receptor | Germline Mutation | Endocrine Disorder |
|---|---|---|
| ACTH/melanocortin-2 receptor | Inactivating mutations (homozygous, compound heterozygous) | Familial glucocorticoid deficiency type 1 |
| Melanocortin-4 receptor | Inactivating mutations (most heterozygous, some homozygous) | Obesity |
| V2 vasopressin receptor | Inactivating mutations (most X-linked recessive, rarely X-linked dominant) | X-linked nephrogenic diabetes insipidus |
| LH receptor | Inactivating mutations (homozygous, compound heterozygous) Activating mutations (heterozygous) | Males: types I and II Leydig cell hypoplasia Females: asymptomatic or hypergonadotropic hypogonadism with primary amenorrhea Males: male limited precocious puberty |
| FSH receptor | Inactivating mutations (homozygous, compound heterozygous) | Females: autosomal recessive hypergonadotropic ovarian dysgenesis or milder hypergonadotropic hypogonadism Males: variable impairment of spermatogenesis |
| TSH receptor | Inactivating mutations (most homozygous or compound heterozygous, rarely heterozygous) Activating mutations (heterozygous) | Resistance to TSH Autosomal-dominant inherited nonautoimmune hyperthyroidism/toxic adenomas |
| GnRH receptor | Inactivating mutations (homozygous or compound heterozygous) | Isolated hypogonadotropic hypogonadism |
| TRH receptor | Inactivating mutations (compound heterozygous) | Central hypothyroidism |
| GPR54 | Inactivating mutations (homozygous, compound heterozygous) | Normosmic isolated hypogonadotropic hypogonadism |
| Ghrelin | Inactivating mutations (homozygous, possible heterozygous) | Short stature because of decreased growth hormone secretion |
| GHRH receptor | Inactivating mutations (homozygous/compound heterozygous) | Isolated growth hormone deficiency |
| Type 1 PTH receptor | Inactivating mutations (homozygous, heterozygous) Activating mutations (heterozygous) | Blomstrand chondrodysplasia if homozygous and rarely if heterozygous; enchondromatosis if heterozygous Jansen metaphyseal chondrodysplasia |
| Calcium-sensing receptor | Inactivating mutations (heterozygous, homozygous) Activating mutations (heterozygous) | Familial benign hypocalciuric hypercalcemia typical if heterozygous, neonatal severe hyperparathyroidism rarely if heterozygous, typical if homozygous Autosomal-dominant hypocalcemic hypocalciuria, Bartter syndrome type V |
The GPCR superfamily is divided into eight major classes. These receptors contain an amino-terminal extracellular domain that is frequently called the ectodomain or exodomain . These receptors also contain seven putative transmembrane spanning alpha helices (TM-I to TM-VII). The alpha helices are connected by three intracellular (i1–i3) and three extracellular (e1–e3) loops that are often collectively called the serpentine region ( Fig. 3.1 ). The carboxy-terminal intracellular region is usually referred to as the endodomain.
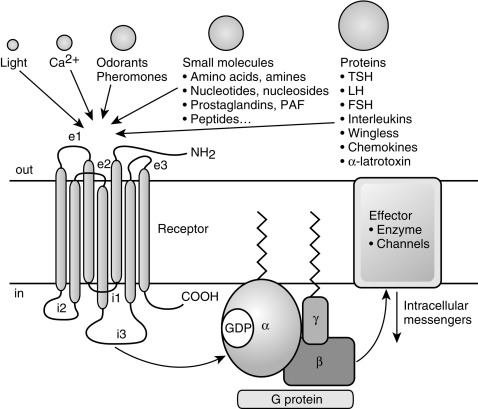
GPCRs are activated by a wide variety of signals, including proteins, nucleotides, amino acid residues, Ca 2 + , light photons, and odorants (see Fig. 3.1 ). It is postulated that ligand binding alters the conformation of transmembrane domains and intracellular loops, increasing the affinity of the receptor for specific heterotrimeric guanosine nucleotide binding proteins (G proteins) (see Fig. 3.1 ). G proteins share a common heterotrimeric structure consisting of an α subunit and a tightly coupled βγ dimer. The α subunit interacts with detector and effector molecules, binds guanosine 5’-triphosphate (GTP), and possesses intrinsic GTPase activity. There are 16 genes in mammals that encode some 20 different α chains. The Gα subunits are categorized in four classes and include Gsα (G stimulatory), Giα (G inhibitory) and Goα (G other), Gq/11α, and G12/13α. They behave differently in the recognition of the effector but share similar structures and mechanism of activation. The Gα subunits consist of two domains: a GTP-binding domain and a helical insertion domain. The GTP-binding domain is homologous to Ras-like small GTPases and includes switch regions I and II, which change conformation during activation. The switch regions are loops of alpha helices with conformations sensitive to guanine nucleotides. The helical insertion domain is inserted into the GTP-binding domain before switch region I and is unique to heterotrimeric G proteins. This helical insertion domain sequesters the guanine nucleotide at the interface with the GTP-binding domain and must be displaced to enable nucleotide dissociation.
The α subunits associate with a smaller group of β(5) and γ(12) subunits. Combinatorial specificity in the associations between various G protein subunits provides the potential for enormous diversity and may allow distinct heterotrimers to interact selectively with only a limited number of GPCRs and effector proteins.
There are two principal signal transduction pathways involving the GPCRs: the cyclic AMP signal pathway and the phosphatidylinositol signal pathway. G protein–induced signal generation is regulated by a “molecular timer” that is determined by the rate of GTP exchange and hydrolysis. In the inactive state, G proteins exist in the heterotrimeric form with guanosine 5’-diphosphate (GDP) bound to the α chain. Interaction of a ligand-bound receptor with a G protein leads to release of GDP, with subsequent binding of GTP to the α chain. The binding of GTP to the α chain leads to dissociation of the α chain from the βγ dimer, allowing the now free α-GTP chain to interact with target enzymes and ion channels. The βγ dimers also participate in downstream signaling events through interaction with an everwidening array of targets, including certain forms of adenylyl cyclase and phospholipase C, potassium channels, and GPCR kinases.
G protein signaling is terminated by the hydrolysis of α-GTP to α-GDP by an intrinsic GTPase. A group of proteins, called regulators of G protein signaling (RGSs), acts as GTPase-activating proteins (GAPs), specific for Gα subunits. These proteins accelerate hydrolysis of GTP to GDP and terminate the transduced signal. In some cases, the effector itself may possess intrinsic GAP activity, which helps deactivate the pathway. This is true in the case of phospholipase C β, which possesses GAP activity within its carboxy-terminal region. This is an alternate form of regulation for the Gα subunit. However, it should be noted that the GAPs do not have catalytic residues to activate the Gα protein. Rather, GAPs reduce the required activation energy for the reaction to take place. After hydrolysis of GTP, the Gα-GDP chain reassociates with the βγ dimer; the reassociated heterotrimeric G protein is now capable of participating in another cycle of receptor-activated signaling.
Specificity in ligand binding is conferred by variations in the primary structures of the extracellular and intracellular domains. Specificity of effector responses is conferred by the variations in the primary structure of intracellular domains and isoforms of the Gα subunits of G proteins. Some GPCRs couple predominantly with Gα i /Gα o subunits that act primarily to decrease adenylyl cyclase activity. Other GPCRs couple predominantly with Gα s subunits that increase adenylyl cyclase activity or Gα q /Gα subunits that increase phospholipase C activity.
Interestingly, data show that cytoskeletal proteins may modulate receptor–G protein coupling. For example, the erythrocyte membrane cytoskeletal protein 4.1G can interfere with A1 adenosine receptor signal transduction. 4.1G also influences metabotropic glutamate receptor 1α-mediated cyclic AMP accumulation, increases the ligand-binding ability of metabotropic glutamate receptor 1α, and alters its cellular distribution. 4.1G may also play a role in receptor-receptor dimerization.
Receptor agonist-independent and agonist-induced homo- and heterodimerization have increasingly been recognized as important determinants of GPCR function. For example, the GPCR somatostatin receptor 5 (SSTR5) primarily exist as monomers in the absence of an agonist. However, they form homodimers in the presence of an agonist. Furthermore, it has been shown that SSTR5 can form heterodimers with type 2 dopamine receptors (DRD2)—another GPCR—in the presence of hsst2 agonist or dopamine. Agonist-induced activation of SSTR5-DRD2 heterodimers in Chinese hamster ovary (CHO) cells expressing SSTR5 and DRD2 is increased, when compared with agonist-induced activation of monomers and homodimers in CHO cells expressing only SSTR5 or DRD2. Heterodimerization of receptors may also lead to inactivation of one of the receptors in the complex. For example, heterodimerization of somatostatin receptor 2A (sst2A) with somatostatin receptor 3 (SSTR3) appears to lead to inactivation of the heterodimerized SSTR3, without inactivating the heterodimerized SSTR2.
GPCRs can form heterodimers with nonreceptor transmembrane proteins. Both the calcitonin receptor (CALCR) and the calcitonin receptor-like protein (CALCRL) can form heterodimers with three different accessory proteins that are termed “ RAMPs ”: RAMP1, RAMP2, and RAMP3. Whereas CALCRs can be activated by ligand in the absence of heterodimerization with a RAMP, CALCRLs are only activated by ligand if heterodimerized with a RAMP. RAMPs alter the ligand specificity of the heterodimerized receptor.
CALCRs that are not in heterodimers with RAMPS are activated by calcitonin and thus constitute the classic CALCR. However, CALCRs heterodimerized with RAMP1, RAMP2, and RAMP3 bind amylin and constitute amylin1, amylin2, and amylin3 receptors, respectively. CALCRLs dimerized with RAMP1 bind calcitonin gene–related peptide and constitute the calcitonin gene–related peptide receptor. CALCRLs dimerized with RAMP2 and RAMP3 bind adrenomedullin and constitute adrenomedullin1 and adrenomedullin2 receptors, respectively. RAMPS alter function of other GPCRs that transduce hormone action. The distribution and function of parathyroid hormone 1 and 2 receptors are altered by binding to RAMP2 and RAMP3, respectively. The distribution and function of the glucagon receptor is altered by binding to RAMP2. Dimerization/heterodimerization may occur in the endoplasmic reticulum (ER), shortly after protein synthesis occurs. The ER plays a role in determining whether or not a protein will be expressed elsewhere in the cell, thus protecting the cell from misfolded and (likely) mutant proteins. The nonheterodimerized CALCRL is an orphan receptor because the CALCRLs cannot leave the ER for the cell membrane, unless heterodimerized with RAMPs.
The melanocortin receptors also use accessory proteins. Circulating adrenocorticotropin hormone (ACTH) binds to five different forms of the melanocortin receptor (types 1–5), but only the melanocortin 2 receptor (MC2R) in the adrenal cortex leads to release of adrenal steroids. MC2R interacts with Gs, which leads to activation of adenylyl cyclase and formation of cyclic AMP. The MC2R is the smallest GPCR known to date and belongs to a family of melanocortin receptors (types 1–5) that bind to various derivatives of proopiomelanocortin, especially α-melanocyte-stimulating hormone (α-MSH). The accessory protein melanocortin 2 receptor-associated protein (MRAP) is required for MC2R function, as it is critical for the translocation of the receptor from the ER to the cell surface. Moreover, MRAP facilitates signaling of the MC2R. Loss of function of MRAP thus prevents membrane expression of MC2R and completely prevents ACTH signaling. MRAP-deficient mice die at birth, unless rescued with glucocorticoids, but have normal mineralocorticoid and catecholamine production. The adrenal glands of MRAP-deficient adult mice are small with abnormal adrenal morphology, abnormal cortex zonation, and abnormal adrenal progenitor cell differentiation. Interestingly, MRAP forms a unique antiparallel homodimer in close proximity to the MC2R. The MRAP accessory protein can also interact with other melanocortin receptors, particularly MC5R, but exerts negative effects on their signaling. Expression of MRAP was shown to be predominantly present in the zona fasciculata in the rat adrenal gland, consistent with its facilitating role in glucocorticoid production. Hence mutations in MC2R or MRAP can lead to familial glucocorticoid deficiency secondary to ACTH resistance. In contrast, MRAP2, a protein with 39% amino acid homology to MRAP, shares the MC2R-trafficking function of MRAP but does not appear to play a major supportive role in adrenocortical ACTH signaling. On the contrary, in vitro studies have shown that overexpression of MRAP2 can suppress MC2R activation. MRAP2 appears to play a role in energy homeostasis as MRAP2 KO mice develop obesity. MRAP2 interacts with MC4R in the paraventricular nucleus of the hypothalamus (PVN) where PVN-specific MRAP2 KO duplicates the global MRAP2 KO phenotype.
Failure of the ER to export mutant GPCR homodimers and mutant GPCR wild-type GPCR heterodimers to the cell membrane has been found to be the cause of dominant negative endocrine conditions. A dominant negative mutation is a heterozygous mutation that results in a phenotype that would be expected by a loss of function in both alleles. Some heterozygous MC4R mutations cause dominantly inherited obesity because of interaction of wild-type MC4R with the mutant receptor, and this specific effect of protein-protein interaction results in a dominant-negative effect. In addition, some heterozygous mutations in the gene encoding the V2 vasopressin receptor cause nephrogenic diabetes insipidus via production of mutant proteins that interfere with transit of normal receptors to the cell’s membrane. These mutant receptors interfere with cell-surface expression of wild-type receptors by forming heterodimers with the wild-type receptors that cannot be exported from the ER to the cell membrane. This finding explains why females heterozygous for these V2 vasopressin receptor gene mutations do not concentrate their urine with even high doses of desmopressin, a synthetic V2 vasopressin receptor agonist, in spite of being able to produce wild-type V2 vasopressin receptors. A similar phenomenon explains dominant transmission of partial thyroid-stimulating hormone (TSH) receptor resistance in patients heterozygous for some inactivating TSH receptor mutations. In these patients, mutant TSH receptors form oligomers with wild-type receptors and prevent export of wild-type receptors from the ER to the cell membrane.
Similarly, misfolding and misrouting of some mutant gonadotropin-releasing hormone (GnRH) receptors in the ER (as well as oligomerization of these mutant GnRH receptors with wild-type GnRH receptors) decrease cell membrane expression of wild-type GnRH receptors. This phenomenon, however, has not been found to have clinical implications in patients who are heterozygous for mutations that cause autosomal recessive isolated hypogonadotropic hypogonadism (IHH), as the heterozygous individuals demonstrate an intact GnRH-gonadotropin axis and do not have clinical signs of IHH. Thus in these individuals, enough wild-type GnRH receptors do not oligomerize with mutant GnRH receptors and are transported to the cell membrane to maintain sufficiently normal GnRH-GnRH receptor interactions to avoid development of IHH.
Most GPCRs activate G proteins at very low levels in the absence of ligand binding. Some GPCRs have much higher constitutive (i.e., ligand-independent) activity, such as luteinizing hormone, TSH, thyrotropin-releasing hormone (TRH), glucagon-like peptide-1, melanocortin, and cannabinoid receptors. These receptors can activate G proteins in the absence of ligand binding, demonstrating constitutive activity that increases linearly with increased cell-surface expression of the receptors. As described earlier, inverse agonists decrease the activity of these receptors. In receptors without constitutive activity, genetic mutations that lead to substitution of a single amino acid can also greatly increase the interaction rate of the unliganded receptor for its G protein. It is possible that inverse agonists may play a role in treating medical conditions caused by GPCR mutations that lead to increased constitutional activation of the receptor.
Receptor desensitization and resensitization play a role in GPCR activity. Three processes for receptor desensitization have been described. The first receptor desensitization process is rapid uncoupling of the G protein from GPCRs. This process occurs within seconds to minutes after initiation of the process and occurs as a result of phosphorylation of GPCRs. G protein receptor kinases (GRKs) have been increasingly recognized as playing a major role when this process involves homologous desensitization. GRK-mediated phosphorylation of serine and threonine residues in the third intracellular loop, or the carboxy-terminal intracellular domain leads to activation of β-arrestins, which in turn inactivate adenylyl cyclase ( Fig. 3.2 ). Second-messenger–dependent protein kinases also contribute to receptor desensitization when this process involves homologous desensitization, but they also participate in receptor desensitization when desensitization involves heterologous desensitization. Heterologous or agonist-independent desensitization occurs as a result of activation of a different receptor from the one that is desensitized.

The second receptor desensitization process is internalization/sequestration of GPCRs. This process is slower than receptor phosphorylation-induced uncoupling of the G protein from GPCRs and occurs within minutes to hours after initiation of the process. In addition to phosphorylation, both the GPCR and β-arrestins are modified posttranslationally in several ways, including by ubiquitination. This process is reversible because the receptors can be recycled to the cell surface (see Fig. 3.2 ). GRKs and β-arrestins play a role in initiating internalization/sequestration of β2-adrenergic, luteinizing hormone (LH), FSH, TSH, TRH, vasopressin V2, angiotensin II type 1A, and other GPCRs in clathrin-coated vesicles (see Fig. 3.2 ). Dephosphorylation of the sequestered receptor, followed by disassociation of the receptor from β-arrestin, is necessary for the receptor to be recycled to the cell membrane and resensitized (see Fig. 3.2 ).
The third receptor desensitization process is downregulation. With downregulation, the number of intracellular GPCRs decreases because of increased lysosomal degradation and decreased synthesis of the receptors caused by alteration of transcriptional and posttranscriptional regulatory mechanisms (see Fig. 3.2 ). Downregulation is a slow process that occurs within several hours to days after initiation of the processes that lead to its development. For many GPCRs, receptor ubiquitination promotes degradation of agonist-activated receptors in the lysosomes. Other proteins also play important roles in desensitization, including phosphodiesterases, RGS family proteins, and A-kinase-anchoring proteins. Together, this intricate network of kinases, ubiquitin ligases, and adaptor proteins orchestrate the acute and prolonged desensitization of GPCRs.
One of the ways the Arg137His V2 vasopressin receptor mutation interferes with mutant receptor function and causes X-linked nephrogenic diabetes insipidus is by altering desensitization and recycling of the mutant receptor. In vitro studies have revealed that the mutant receptor is constitutively phosphorylated. Thus even in the absence of ligand binding, the mutant receptor is bound by β-arrestin—which in turn leads to sequestration of the mutant receptor within clathrin-coated vesicles. Recycling of the mutant receptor back to the cell membrane requires the mutant receptor to be dephosphorylated and disassociated from β-arrestin. However, the mutant receptor does not undergo dephosphorylation, while sequestered, and thus cannot be disassociated from β-arrestin and recycled to the cell membrane—thereby reducing cell membrane expression of the mutant receptor.
Of the eight classes of GPCRs, only classes A, B, and C contain receptors for mammalian hormones and neurotransmitters ( Fig. 3.3 ). Class A receptors contain the rhodopsin-like receptors and are divided into at least 15 groups. Four of these groups contain receptors activated by hormones. These are the peptide receptor, hormone protein receptor, GnRH receptor, and the TRH and secretagogue receptor groups.
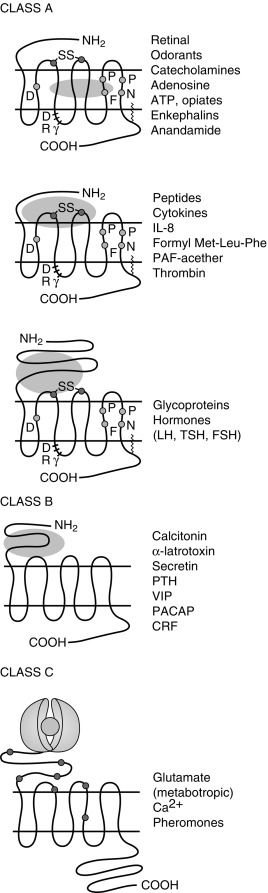
The peptide receptor group includes the angiotensin, ACTH/melanocortin, oxytocin, somatostatin, and vasopressin receptors. The hormone protein receptor group includes the receptors for glycoprotein hormones, including FSH, LH, and thyrotropin (TSH) receptors. These receptors have large extracellular amino-terminal domains and ligand-binding sites that include the first and third extracellular loops (see Fig. 3.3 ). There is also much similarity in amino acid sequence among these receptors (see Fig. 3.3 ). The GnRH receptor group only contains the GnRH receptor. The TRH and secretagogue receptor group includes the TRH receptor and the growth hormone (GH) secretagogue receptor.
Class B GPCRs are structurally similar to members of the hormone protein receptor group (see Fig. 3.3 ). However, unlike the glycoprotein hormone receptors, class B GPCRs do not share similar amino acid sequences. This family contains receptors for higher molecular weight hormones, including calcitonin, glucagon, gastric inhibitory peptide, parathyroid hormone (PTH), and corticotrophin-releasing factor (CRF).
Class C receptors have a large extracellular domain, with two lobes separated by a hinge region that closes on the ligand (see Fig. 3.3 ). This region has also been called the Venus flytrap domain or module because of the trapping mechanism of the hinge region. This family includes the calcium-sensing receptor (CASR).
Most GPCR-inactivating mutations can be classified into one of five classes. Class I inactivating mutations interfere with receptor biosynthesis. Class II inactivating mutations interfere with receptor trafficking to the cell surface. Class III inactivating mutations interfere with ligand binding. Class IV inactivating mutations impede receptor activation. Class V inactivating mutations do not cause discernible defects in receptor biosynthesis, trafficking, ligand binding, or activation, but may cause medical disorders. There are also inactivating mutations that interfere with receptor function via multiple mechanisms and thus cannot be placed into one class.
Class A receptors that transduce hormone action
The Peptide Receptor Group
Adrenocorticotropin and Melanocortin-2 Receptors
An alternative name for the ACTH receptor is melanocortin-2 receptor (MC2R) because the ACTH receptor is one of five members of the melanocortin receptor family of GPCRs, all of which couple to Gs to activate generation of intracellular cyclic AMP. For the purpose of clarity, when discussing interactions between ACTH and its receptor, the older name will be used for the remainder of this chapter. The ACTH receptor gene is located on small arm of chromosome 18 (18p11.2). The ACTH receptor has a small extracellular and intracytoplasmic domain. Adrenocorticotropin-induced activation of the ACTH receptor in the zona fasciculata and zona reticularis of the adrenal cortex stimulates G s , resulting in increased intracellular cyclic AMP levels that stimulate steroidogenesis by activating cyclic AMP-dependent kinases.
Hereditary isolated glucocorticoid deficiency, resistance to ACTH, and familial glucocorticoid deficiency (FGD) are the same names for an autosomal recessive syndrome that consists of glucocorticoid deficiency accompanied by normal mineralocorticoid secretion. FGD has been classified further as FGD types 1 and 2 and the triple A syndrome. Patients with FGD type 1 have biallelic MC2R mutations, resulting in ACTH receptors with abnormal function, and account for 25% of FGD cases. In contrast, patients with FGD type 2 have ACTH resistance caused by mutations in MRAP. Triple A (Allgrove syndrome) is an autosomal recessive syndrome characterized by ACTH-resistant adrenal insufficiency, achalasia, and alacrima—which is caused by mutations in the achalasia-adrenocortical insufficiency-alacrima syndrome ( AAAS ) gene encoding the protein ALADIN. ALADIN is thought to regulate nuclear pore complexes, and nucleocytoplasmic transport plays a role in modulating the oxidative stress response in adrenal cells.
The spectrum of disease in patients with FGD type 1 varies from presentation as a neonate with hypoglycemia, seizures, or circulatory collapse to presentation in childhood with fatigue and increased susceptibility to infection. Less commonly, patients may present with childhood asthma that resolves with treatment with physiologic doses of glucocorticoids. Hyperpigmentation thought to be caused by increased ACTH levels acting on the MC1R may be seen as early as the first month of life, but usually becomes apparent after the fourth month of life. There is one reported case of FGD type 1 without hyperpigmentation, despite elevated ACTH levels, in a patient with homozygous mutations in both the MC2R and the MC1R. The assumption that hyperpigmentation is caused by increased ACTH levels acting on the MC1R (both in FGD type 1 and Addison disease) was substantiated in this patient whose MC1R mutation had previously been implicated in red hair and pale skin phenotypes. Neonates with FGD type 1 may also suffer from jaundice. Tall stature accompanied by an advanced or dissociated bone age, in spite of normal age of onset of puberty, appears to be common in children with FGD type 1. The pathophysiology of tall stature has not been definitively elucidated. One theory is that the anabolic effects of GH are unopposed by cortisol.
Patients with FGD1 exhibit absent adrenarche, confirming the importance of ACTH in the induction and maintenance of adrenarche. At presentation, plasma cortisol, androstenedione, and dihydroepiandrosterone levels are low or low normal—and plasma ACTH levels are elevated. When supine, patients with FGD type 1 have renin and aldosterone levels that are near normal. Histologically, the zona fasciculata and zona reticularis are atrophied with FGD. However, demonstrating the lack of an essential role for ACTH in the embryologic development and maintenance of the zona glomerulosa, adrenal cortices in patients with all types of FGD contain zona glomerulosa cells.
FGD type 2 is caused by mutations in MRAP1 as described earlier and accounts for 20% of all FGD cases. Patients with FGD type 2 present with severe symptoms at an earlier age than patients with FGD type 1 (median age 0.08 years vs. 2 years). Patients can present with severe neurologic disability, seizures, and microcephaly thought to be caused by unrecognized hypoglycemia. Patients with FGD type 2 have normal heights. This is thought to be caused by early treatment with glucocorticoids. Mutations are splice site or nonsense mutations predicted to produce proteins without the transmembrane domain, which is necessary for interaction with MC2R.
Abnormalities in ACTH receptor expression may be seen in other conditions. Evidence suggests that the ACTH receptor-Gα s -adenylyl cyclase-cyclic AMP cascade maintains differentiation of adrenocortical cells and that impairment of this cascade leads to dedifferentiation and increased proliferation of adrenocortical cells. Adrenocortical carcinomas from some patients have been found to have a loss of heterozygosity (LOH) for the ACTH receptor gene, resulting in markedly decreased ACTH receptor messenger ribonucleic acid (mRNA) expression. Growth of the tumors with LOH for the ACTH receptor gene also may be more aggressive than the other tumors. An activating mutation of G i2 that constitutively suppresses adenylyl cyclase activity has also been found in adrenocortical tumors. Thus decreased ACTH receptor activity may be associated with tumorigenesis.
Interestingly, many patients with ACTH-independent macronodular adrenal hyperplasia (AIMAH)—a cause of ACTH-independent Cushing syndrome caused by inactivating mutations of the putative tumor-suppressor gene ARMC5 —exhibit increased glucocorticoid levels in response to noncorticotropin hormones that do not normally induce glucocorticoid release. These hormones include gastric inhibitory peptide, exogenous arginine and lysine vasopressin, LH, human chorionic gonadotropin (HCG), angiotensin II, catecholamines, leptin, and serotonin receptor agonists. Increased expression of the receptors for these ligands in the abnormal adrenal glands has been implicated as a possible explanation for the abnormal induction of glucocorticoid release by these noncorticotropin ligands. However, receptors for some of these ligands are expressed in normal adrenal glands. Thus the mechanism for this phenomenon remains to be fully elucidated.
Other Melanocortin Receptors
Murine studies reveal that melanocortin-3 receptor (MC3R), another of the five members of the melanocortin receptor family, regulates fat deposition, as mice with MC3R deficiency or partially inactive receptors have normal resting energy expenditure and increased fat mass and reduced fat-free mass, including decreased bone formation. The role of the MC3R in humans is less clear. More than 24 human MC3R variants have been identified without evidence of obesity. However, patients with these variants were not phenotyped for fat mass to assess whether they phenocopy the mouse model, which exhibits normal body weight but greater energy intake and altered energy partitioning that is biased toward lipid-accumulating cells’ altered partitioning. Several coding variants (p.D158Y, p.T280S, p.I183N), which are likely pathologic because of significantly decreased cyclic AMP production, as well as noncoding variants, demonstrate early-onset obesity in all affected individuals. Homozygosity for a pair of single-nucleotide polymorphisms of the MC3R gene (p.T6K + p.V81I) that result in production of partially inactive MC3Rs was found to be associated with pediatric-onset obesity in Caucasian American and African American children. Subjects homozygous for the double mutation had higher body mass index (BMI)-z, fat mass and percent fat mass, as well as waist circumference. That is despite finding no differences in energy intake, resting energy expenditure, total energy expenditure, respiratory quotient, physical activity. A mouse model carrying the human MC3R T6K + V81I variants showed that the double mutant exhibited greater fat mass and feeding efficiency with reduced fat-free mass. These findings are similar to the MC3R knockout mouse.
The melanocortin-4 receptor (MC4R) is another member of the melanocortin receptor family and plays a role in controlling appetite and weight. The MC4R has baseline constitutive (i.e., ligand-independent) activity that can be inhibited by the inverse agonist agouti-related peptide (AgRP). Activation of the MC4R by its natural agonist α-MSH produces anorexigenic effects. More than 150 naturally occurring MC4R mutations have been identified, causing hyperphagic obesity, increased lean body mass, increased bone density, and increased linear growth. Patients with homozygous mutations appear to have more severe obesity than their heterozygous relatives, consistent with codominant inheritance.
MC4R mutations are thought to be the most common monogenic cause of human obesity. The prevalence of pathogenic MC4R mutations in obese populations varies widely, ranging from 0.5% to 5.8%, depending on the screening criteria and population. AgRP gene polymorphisms appear to be associated with anorexia nervosa.
Little is known about melanocortin-5 receptors (MC5Rs) in animals and humans. There is only weak evidence from a single linkage and association study of families in Quebec that suggests that MC5Rs may also play a role in regulating body weight and fat mass.
Another member of the melanocortin receptor family, the MC1R, controls skin and hair pigmentation. Activation of MC1Rs in skin and hair follicle melanocytes by the proopiomelanocortin (POMC)-derived peptides α-MSH and ACTH stimulates the synthesis of eumelanin, a brown-black pigment. Inhibition of MC1R baseline constitutive activity by agouti protein, or specific mutations, leads to release of pheomelanin, a red-yellow pigment, from the melanocytes.
Inactivating homozygous mutations of the POMC gene cause hypoadrenalism, red hair, fair skin, and early-onset obesity. Hypoadrenalism is characterized by glucocorticoid deficiency because of lack of ACTH production from the POMC precursor. Fair skin and red hair are caused by a lack of ACTH and α-MSH-induced melanocyte release of eumelanin that results from activation of MC1Rs. Of note, nonwhite patients with homozygous POMC mutations do not appear to have the fair skin and red hair phenotype. In white individuals, eumelanin synthesis appears to be dependent on POMC-derived peptides, whereas in darker individuals, other genes may control eumelanin synthesis. Obesity is caused by lack of α-MSH–induced anorectic effects, which normally result when α-MSH activates MC4Rs. Heterozygosity for POMC gene mutations has been associated with hyperphagia, early-onset obesity, and increased linear growth. Both homozygous and heterozygous mutations of prohormone convertase 1 cause obesity in humans. Prohormone convertase 1 acts on POMC, proinsulin, and proglucagon. Patients with prohormone convertase 1 deficiency also have neonatal enteropathy and postprandial hypoglycemia. The cause of enteropathy is unknown but hypothesized to be related to the processing of GLP-2 by prohormone convertase 1. GLP-2 is known to stimulate proliferation and repair of intestinal epithelium.
Vasopressin Receptors
Nephrogenic diabetes insipidus (NDI) results from decreased responsiveness of the renal tubule to arginine vasopressin (AVP), with resulting excessive loss of free water. NDI is characterized by polydipsia and polyuria that is not responsive to vasopressin and vasopressin analogs. Vasopressin binds to the V2 vasopressin receptor (AVPR2), a Gs-coupled receptor, in the basolateral membrane of collecting duct principal cells in the kidney and activates translocation of aquaporin-2 (AQP2) water channels to the apical membrane, thereby inducing water permeability. X-linked NDI is caused by inactivating mutations of the V2 vasopressin receptor ( AVPR2 ) gene located at Xq28 and accounts for about 90% of genetically determined NDI. More than 200 AVPR2 mutations have been described, including missense, nonsense, insertions, deletions, and complex rearrangements. Mutations have been categorized into five classes based on mechanism, including abnormal transcription, mRNA processing, translation, aberrant folding and intracellular retention, loss of the G protein binding site, loss of the AVP binding site, and defects in intracellular trafficking. Some patients with X-linked NDI are responsive to high doses of desmopressin. Autosomal recessive NDI (ARNDI) is caused by loss-of-function mutations in the gene for the AQP2 water channel and accounts for about 10% of genetic forms of NDI. More than 50 known mutations cause ARNDI. Autosomal dominant forms of NDI are also caused by mutations in AQP2 that are functional, but fail to be transported to the apical membrane. Eleven mutations have been described accounting for less than 1% of genetic forms of NDI and generally have a milder phenotype than ARNDI or X-linked NDI.
Gain-of-function mutations in the V2 vasopressin receptor have also been reported. Deoxyribonucleic acid (DNA) sequencing of two patients’ V2R gene identified heterozygous missense mutations in both, with resultant changes in codon 137 from arginine to cysteine (p.R137C) or leucine (p.R137L). These mutations resulted in constitutive activation of the receptor and clinical features of inappropriate antidiuretic hormone secretion (SIADH), which was termed nephrogenic syndrome of inappropriate antidiuresis (NSIAD) . To date, around 30 cases of NSIAD have been reported, most of which are described in males as the condition is X-linked. Patients can present in infancy but sometimes do not present until adulthood. There have been several reports of female patients with NSIAD, some diagnosed in infancy and others in adulthood. Patients with the p.R137L mutation demonstrated the expected decrease in AVP levels with a water-loading test, but urine AQP2 levels remained inappropriately elevated.
The Glycoprotein Hormone Receptor Group
The glycoprotein hormones include TSH, FSH, LH, and HCG. These hormones share common α subunits that dimerize with hormone-specific β subunits. TSH, FSH, and LH bind to the extracellular amino-terminal domain of the TSH, FSH, and LH receptors, respectively. The effects of HCG are mediated by the LH receptor, which is also known as the luteinizing hormone/choriogonadotropin receptor ( LHCGR ).
Glycoprotein hormone receptors have a large (350 to 400 residues) extracellular amino-terminal domain, also known as the ectodomain , that participates in ligand binding (see Fig. 3.3 ). The ectodomain includes leucine-rich repeats that are highly conserved among the glycoprotein hormone receptors. There is 39% to 46% similarity of the ectodomain and 68% to 72% similarity of the transmembrane or serpentine domain among the three glycoprotein hormone receptors.
The glycoprotein hormone receptors are coupled to G s , and hormone binding stimulates adenylyl cyclase, leading to increased intracellular cyclic AMP levels and protein kinase A (PKA) activation. Mutations leading to endocrine dysfunction have been reported for each of the glycoprotein hormone receptors.
Luteinizing Hormone/Choriogonadotropin Receptors
Both inactivating and activating mutations of the LH receptor have been found in humans. The LH receptor gene is located in chromosome 2p21 and consists of 11 exons. Exon 1 encodes a peptide that directs the LH receptor to the plasma membrane. Exons 2 through 10 encode the ectodomain. The last exon encodes the transmembrane domains that are also known as the serpentine regions . Single nonsense mutations, amino acid changes, and partial gene deletions have been described that generate LH receptors with decreased activity. Single–amino-acid changes have also been found that lead to activation of G s in the absence of ligand binding.
Development of LH resistance requires biallelic mutations that inactivate the LH receptor gene, as one normal receptor allele is capable of producing adequate receptor protein to ensure physiologic signaling. In contrast, activating mutations of the LH receptor gene cause endocrine disorders in the heterozygous state.
In the fetus, LH receptors are primarily activated by HCG. Leydig cells begin to express LH receptors shortly after testicular differentiation at 8 weeks of gestation. Thereafter, androgen production, caused by activation of these receptors by HCG, plays an important role in the development of male genitalia and testicular descent. Thus male infants, with inactivating mutations of the LH receptor, may present with abnormally developed genitalia—including micropenis, cryptorchidism, and an XY disorder of sexual differentiation.
Males with mutations that completely inactivate the LH receptor exhibit failure of fetal testicular Leydig cell differentiation. This phenotype, which is known as type 1 Leydig cell hypoplasia , includes female external genitalia with a blind-ending vagina, absence of Müllerian derivatives, and inguinal testes with absent or immature Leydig cells. In addition, patients have elevated serum LH levels, normal serum FSH levels, and decreased serum testosterone levels that do not increase in response to HCG administration. Mutations that lead to this phenotype include a nonsense mutation (Arg545Stop) that results in a receptor that is missing TM4-7, an p.Ala593Pro change, and a TM7 deletion (p.Leu608del, p.Val609) that decreases cell-surface expression of the LH receptor. These mutant receptors are unable to couple to G s .
Males with mutations that do not completely inactivate the LH receptor present with type 2 Leydig cell hypoplasia, which is characterized by a small phallus and decreased virilization. A mutation that leads to this phenotype includes the insertion of a charged lysine at position 625 of TM7 in place of hydrophobic isoleucine that disrupts signal transduction. Another mutation (p.Ser616Tyr, found in patients with mild Leydig cell hypoplasia) is associated with decreased cell-surface expression of the LH receptor. Other deletion and nonsense mutations have also been found to cause mild Leydig cell hypoplasia.
Males with inactivating mutations of the LH receptor may also present with a phenotype intermediate in severity between type 1 and type 2 Leydig cell hypoplasia. A compound heterozygote patient with p.Ser616Tyr on one allele and an inactivating deletion (△exon 8) on the other allele, presented with Leydig cell hypoplasia, micropenis, and hypospadias. The Cys131Arg mutation has also been found in patients with Leydig cell hypoplasia, small phallus, and hypospadias. This mutation is located in the leucine-rich repeat segment of the LH receptor extracellular domain and interferes with high-affinity ligand binding.
Deletion of exon 10 of the LH receptor gene leads to an LH receptor that binds LH and HCG normally. Interestingly, whereas HCG binding can elicit normal transmembrane signaling, LH binding fails to activate the receptor. Because HCG is the principal hormone in utero that activates the LH receptor, and second-messenger response of the mutant receptors to HCG is not impaired, it is not surprising that a male patient found to be homozygous for the mutation was born with normal male genitalia. Pubertal progression and later gonadal function, however, are dependent on LH activation of the LH receptor.
Because deletion of exon 10 of the LH receptor gene results in a mutant LH receptor, with diminished intracellular signaling in response to LH, it is also not surprising that the patient homozygous for this mutation was found to have delayed pubertal development, small testes, and hypergonadotropic hypogonadism, when evaluated at the age of 18 years. Prolonged HCG therapy resulted in normalization of testicular testosterone production, increased testicular size, and the appearance of spermatozoa in semen. Similarly, inactivating mutations of the LH β subunit cause abnormal pubertal development, severe testosterone deficiency, and azoospermia, but normal external genitalia in males. In females, inactivating mutations of the LH β subunit are associated with normal pubertal development and menarche followed by oligomenorrhea, enlarged multicystic ovaries and infertility.
Females with loss of function mutations of the LH receptor may be asymptomatic or present with amenorrhea or oligomenorrhea. Females with complete inactivating LH receptor mutations commonly have primary amenorrhea, inability to ovulate, and decreased estrogen and progesterone levels, accompanied by elevated LH and FSH levels. Affected individuals may have signs of low estrogen levels, including a hypoplastic uterus, a thin-walled vagina, decreased vaginal secretions, and decreased bone mass, although pubertal breast development is normal. Homozygous LH receptor mutations (p.N400S, and p.Ala449Thr) have been associated with empty follicle syndrome, a disorder in which no oocytes are retrieved during in vitro fertilization.
Mutations that constitutively activate LH receptors cause male-limited precocious puberty (MLPP), also known as testotoxicosis —which may be familial or sporadic. Boys with this condition develop GnRH-independent precocious puberty before the age of 4 years, when p.Asp578Gly is present, and as early as the first year of life, when the p.Asp578Tyr mutation is present. Patients with this condition may also have an enlarged phallus at birth.
During the first 5 years of life, patients with MLPP have very low LH and FSH levels but have testosterone levels in pubertal range. During adolescence and adult life, testosterone levels do not increase above age-appropriate concentrations and gonadotropin levels normalize. Thus adolescents and adults with MLPP do not usually manifest signs of androgen excess (such as hirsutism or severe acne). Most mutations that cause MLPP are located in the TM6 and i3, regions that participate in receptor-Gs protein coupling. A milder phenotype was reported in a patient with a heterozygous activating mutation (p.C617Y) in TM7. This mutation was inherited from the patient’s mother who was apparently unaffected. Somatic activating mutations cause sporadic Leydig cell adenomas.
Activating mutations of the LH receptor do not appear to cause clinical disturbances in females. In prepubertal girls, this may be caused by low or absent LH receptor expression or because of insufficient aromatase expression in prepubertal granulosa cells. During puberty, activation of LH receptors on ovarian theca cells leads to the production of androgens that are converted to estrogens by aromatase in granulosa cells. LH, along with FSH, also plays a role in inducing the differentiation of follicles into Graafian follicles and triggers ovulation and release of the oocyte. Detailed phenotyping of the carrier mother of an MLPP male with the p.Asp578Gly activating mutation of the LH receptor failed to reveal any abnormalities in her menstrual cycles or fertility. LH dynamics, androgen, and FSH levels, as well as response to GnRH agonists, were normal.
Follicle-Stimulating Hormone Receptors
Inactivating and activating FSH receptor mutations have been described, but they are far less common than LH receptor mutations. The FSH receptor gene is located in chromosome 2 at p21 and contains 10 exons. The last exon of the FSH receptor gene encodes the transmembrane and intracellular domains.
FSH is required in females for normal follicle maturation and regulation of estrogen production by ovarian granulosa cells. FSH is required in pubertal males for Sertoli cell proliferation, testicular growth, and the maintenance of spermatogenesis.
The first inactivating mutation of the FSH receptor was found in Finnish females with autosomal recessive hypergonadotropic ovarian dysgenesis (ODG). ODG is characterized by primary amenorrhea, infertility, and streak or hypoplastic ovaries in the presence of a 46XX karyotype and elevated gonadotropin levels. Twenty-two out of 75 Finnish patients with ODG were found to be homozygous for a c.C566T point mutation in exon 7 of the FSH receptor gene. This mutation leads to the production of an FSH receptor with an p.Ala189Val substitution in an area of the extracellular ligand-binding domain that is thought to play a role in turnover of the receptor or in directing the receptor to the plasma membrane. The mutated receptor demonstrates normal ligand-binding affinity but has decreased binding capacity and impaired signal transduction, when studied in transfected mouse Sertoli cells. Males homozygous for this mutation have variable impairment of spermatogenesis and low to low-normal testicular volume, but are not azoospermic and can be fertile. The c.C556T point mutation is uncommon outside Finland, where the carrier frequency is 0.96%. Other mutations that alter signal transduction but not receptor expression or binding include p.Ala189Val, p.Asn191Ile, p.Ala419Thr, and p.Phe591Ser. The p.Ala189Val mutation causes primary hypergonadotropic amenorrhea in women and no spermatogenesis in men in the homozygous state and secondary amenorrhea in the heterozygous state. The nearby Asn191Ile mutation also causes hypergonadotropic amenorrhea in the homozygous state, but no clinical phenotype in the heterozygous state. The Ala419Thr mutation was identified in a heterozygous woman with primary amenorrhea. The Phe591Ser mutation causes primary amenorrhea and premature ovarian failure (POF) in the homozygous state and a predisposition to sex cord ovarian tumors in the heterozygous state. Primary amenorrhea and POF have been described in women with homozygous mutations that totally impaired receptor binding to FSH or that resulted in reduced expression of the FSH receptor on the cell surface.
A more recently published series described 13 Chinese families with nonsyndromic premature ovarian insufficiency. They found three novel mutations and 11 previously described homozygous mutations and 3 previously described compound heterozygous mutations. Clinical manifestations ranged from primary amenorrhea to normal menarche with oligomenorrhea or secondary amenorrhea. It was the first study to identify a frameshift mutation (p.Lys140Argfs*16 in the ectodomain) and a missense mutation in the signal peptide of the FSH receptor (p.Gly15Asp). The other novel mutation was p.Pro504Ser in the transmembrane domain.
Compound heterozygosity for mutations that cause partial loss of FSH receptor function may cause endocrine dysfunction in women. Women may present with infertility, secondary amenorrhea, osteoporosis, and a history of normal or delayed onset of puberty, accompanied by elevated LH and FSH, low-normal plasma estradiol, low plasma inhibin B levels, slightly enlarged ovaries with immature follicles, and a small uterus. This may be caused by FSH receptor gene mutations that result in an p.Ile160Thr mutation in the extracellular domain that impairs cell-surface expression and an p.Arg573Cys mutation in e3 that interferes with signal transduction. Other women present with primary amenorrhea and very elevated gonadotropin, low plasma estradiol and inhibin B levels, normal-size ovaries with immature follicles, and a normal-size uterus. This condition is associated with an p.Asp224Val substitution in the extracellular domain, leading to impaired cell-surface expression and a p.Leu601Val substitution in e3 impairing signal transduction.
Activating mutations of the FSH receptor have also been described. Surprisingly, a hypophysectomized male was found to be fertile and to have serum testosterone levels above 4.9 nmol/L (141 ng/dL) and normal testis volume, in spite of undetectable gonadotropin levels. This patient was found to be heterozygous for a c.A1700G mutation in exon 10 of the FSH receptor gene that resulted in a p.Asp567Gly substitution in an area of the third intracytoplasmatic loop that is highly conserved among FSH, LH, and TSH receptors. The same substitution in corresponding areas of the LH and TSH receptors also results in constitutively active receptors and is found in MLPP and thyroid adenomas, respectively. Other activating mutations have been identified to cause spontaneous ovarian hyperstimulation syndrome (OHSS). OHSS is a common complication of treatment protocols used to induce ova for in vitro fertilization and is characterized by multiple follicular cysts lined by luteinized cells, which can result in abdominal discomfort and distention, as well as ovarian enlargement and fluid sequestration. One such mutation is the p.Asp567Asn, which was found in a woman with recurrent spontaneous OHSS. The p.Thr449Ile and p.Thr449Ala mutations cause a conformational change that leads to loss of specificity for FSH, leading to sensitivity to HCG and TSH, causing spontaneous OHSS during pregnancy or with hypothyroidism. The p.Ile545Thr mutation caused spontaneous OHSS in a woman during the first trimester of pregnancy, despite a normal HCG level. This mutant receptor displayed detectable constitutive activity, as well as promiscuous activation by HCG and TSH.
Thyroid-Stimulating Hormone Receptors
The TSH receptor gene is located on chromosome 14 and contains 10 exons, with the first nine exons encoding the large extracellular domain and the 10 th exon coding the remainder of the receptor. At low extracellular TSH concentrations, TSH receptor activation leads to stimulation of Gα s —which activates adenylyl cyclase, resulting in increased intracellular cyclic AMP levels. At higher extracellular TSH concentrations, activation of the TSH receptor also stimulates the G q and G 11 proteins—activating phospholipase C and resulting in the production of diacylglycerol and inositol phosphate.
TSH receptors differ from the other glycoprotein hormone receptors in that they exist in two equally active forms. These are the single-chain and two-subunit forms of the TSH receptor ( Fig. 3.4 ). The single-chain form of the TSH receptor is made up of three contiguous subunits: the A subunit, C peptide, and B subunit. The A subunit begins at the amino-terminal of the extracellular domain and contains most of the extracellular domain. The C peptide is connected to the carboxy-terminal of the A subunit and continues the extracellular domain. The C peptide contains a 50-amino-acid sequence that is only found in TSH receptors. The B subunit is connected to the C terminal of the C peptide and contains the TMs and the carboxy-terminal cytoplasmic portion of the receptor. The two-subunit form of the receptor is missing the C peptide, which is cleaved from the protein during intracellular processing and consists of the A and B subunits attached by disulfide bonds. It is surprising that both receptor forms are activated equally by TSH because the C peptide and nearby regions of the A and B subunits participate in signal transduction.
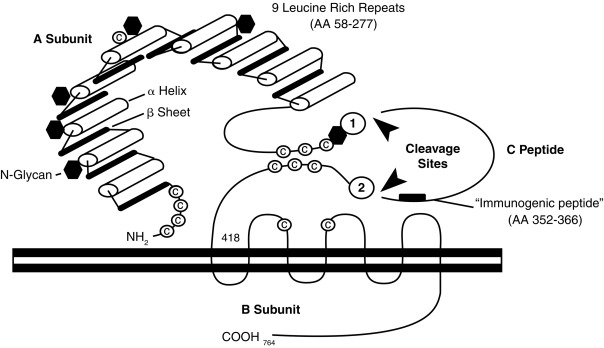
Missense mutations of the TSH receptor gene, leading to replacement of Ser-281 near the carboxy-terminus of the A subunit, with Ile, Thr, or Asn, result in a constitutively active TSH receptor that may cause intrauterine or congenital hyperthyroidism, or toxic adenomas. Activating somatic mutations that cause toxic adenomas have also been found in different transmembrane domains of the TSH receptor. More specifically, clusters of mutations are located in the i3 and TM6 regions—found to be involved with signal transduction in all glycoprotein hormone receptors. The prevalence of activating mutations of the TSH receptor in toxic adenomas has been estimated to range from 2.5% in Japan to 86% in Brazil.
Activating somatic mutations of the TSH receptor have also been found in multinodular goiters. Interestingly, different activating mutations have been found in separate nodules in the same individual. Some well-differentiated thyroid carcinomas have activating mutations of the TSH receptor. Somatic activating mutations of the GNAS gene encoding Gα s have also been found in some toxic adenomas and differentiated thyroid carcinomas. Activating germline mutations of the TSH receptor can cause sporadic or autosomal dominant inherited nonautoimmune hyperthyroidism that presents in utero, during infancy, during childhood, and in some cases in adulthood. These mutations have been found in the amino-terminal extracellular and transmembrane domains.
Patients with heterozygous mutations that lead to constitutively active TSH receptors typically develop hyperthyroidism. In contrast, biallelic loss of function mutations in the TSH receptor genes cause hypothyroidism. Most known loss-of-function TSH receptor mutations are located in the amino-terminal extracellular domain. A spontaneous p.Asp410Asn substitution, near the carboxy-terminus of the C peptide, results in a TSH receptor, with normal ligand binding affinity and impaired Gα s -mediated signal transduction. Patients who are homozygous for this mutation present with compensated hypothyroidism.
Patients homozygous or compound heterozygous for loss-of-function mutations of the TSH receptor present with the syndrome of resistance to TSH (RTSH). Loss-of-function mutations of the TSH receptor that cause RTSH have been identified in the amino-terminal extracellular domain, TM4, TM6, i2, e1, and e3. Clinical severity of RTSH may range from a euthyroid state accompanied by elevated TSH levels (fully compensated RTSH), to mild hypothyroidism unaccompanied by a goiter (partially compensated hypothyroidism), to congenital thyroid hypoplasia accompanied by profound hypothyroidism (uncompensated RTSH). In patients with uncompensated RTSH, a small bilobar thyroid gland is located at the normal site. Loss-of-function mutations of the TSH receptor are a rare cause congenital hypothyroidism more common in Japan and Taiwan (≤ 7% of children), where p.R450H is particularly frequent. Because expression of the sodium-iodide symporter is TSH dependent, thyroid gland uptake of iodine and 99m pertechnetate is diminished or absent in patients with RTSH. In rare cases, iodine uptake is high-normal. These compound heterozygous mutations of the TSH receptor had some Gα s activity and no G q activity, suggesting that iodine uptake is solely controlled by Gα s activity and not G q activity. Some families have been found to have an autosomal-dominant form of RTSH that is not caused by a mutation of the TSH receptor.
Human Chorionic Gonadotropin and Thyroid-Stimulating Hormone Receptors during Pregnancy
Because of its structural similarity with TSH, at very high concentrations, HCG can activate the TSH receptor. During pregnancy, HCG activation of TSH receptors leads to elevation in thyroid hormones after the ninth week of gestation—and decreases in TSH levels between the 9th and 12 th weeks of gestation. This phenomenon does not usually result in maternal hyperthyroidism (gestational thyrotoxicosis). However, when HCG levels are abnormally elevated because of gestational trophoblastic disease caused by a molar pregnancy or choriocarcinoma, hyperthyroidism may occur. The prevalence of thyrotoxicosis in gestational trophoblastic disease correlates with HCG levels. In one study of 196 patients treated with chemotherapy for gestational trophoblastic neoplasia, the prevalence of thyrotoxicosis was 7%. Biochemical thyrotoxicosis only occurred in patients with HCG levels > 10 5 and clinical thyrotoxicosis only occurred in patients with HCG levels greater than 10 6 . Serum TSH is consistently suppressed when HCG levels are above 4 × 10 5 mIU/mL.
A mother and daughter were identified with recurrent gestational hyperthyroidism and normal serum HCG levels. These individuals were found to be heterozygous for a missense mutation in the TSH receptor gene, resulting in a p.Lys183Arg substitution in the extracellular domain of the receptor. It is believed that this substitution increases sensitivity of the receptor to activation by HCG, causing gestational hyperthyroidism.
The Gonadotropin-Releasing Hormone Receptor Group
Gonadotropin-Releasing Hormone Receptors
The GnRH receptor gene is located on 4q13 and includes three exons. Unlike glycoprotein hormone receptors, GnRH receptors lack an intracellular carboxy-terminal domain. In contrast to most GPCRs, the GnRH receptor is coupled to G q /G 11 and hence ligand-binding leads to stimulation of phospholipase C and not adenylyl cyclase. Phospholipase C cleaves phosphatidylinositol-4,5-diphosphate (PIP2) to inositol 1,4,5-triphosphate (IP3) and diacylglycerol, leading to increased protein kinase C activity.
Some patients with IHH are homozygous or compound heterozygous for loss-of-function mutations in the GnRH receptor gene. Unlike patients with Kallmann syndrome (KS), they have a normal sense of smell. GnRH receptor mutations were found in approximately 5% of patients with normosmic congenital hypogonadotropic hypogonadism. GnRH receptor mutations that cause IHH result in decreased binding of GnRH or impaired GnRH receptor signal transduction, or decreased GnRH receptor cell membrane expression because of misrouting of GnRH receptor oligomers from the ER. Some mutations, including p.E90K, p.L266R, and p.S168R, that cause misfolding and retention, within the ER, exhibit a dominant negative effect because of retention of wild-type receptors.
Female patients with mutations that partially compromise GnRH receptor function may present with primary amenorrhea and infertility, associated with a normal breast development, normal or small uterus, and small ovaries with immature follicles. Males with the same mutations may present with incomplete hypogonadotropic hypogonadism (characterized by a delayed and incomplete puberty) or with complete hypogonadotropic hypogonadism (characterized by absent puberty).
Some patients with IHH caused by mutated GnRH receptors have partial or normal gonadotropin responses to exogenous GnRH. However, decreased amplitude in the pulsatile LH secretion can be observed in these patients. Females with a partial or normal gonadotropin response to exogenous GnRH are more likely than nonresponders to become fertile in response to pulsatile exogenous GnRH.
Activating mutations of the GnRH receptor have not been described in the germline or in pituitary adenomas.
The Thyrotropin-Releasing Hormone and Secretagogue Receptor Group
Thyrotropin-Releasing Hormone Receptors
Like the GnRH receptor, TRH receptor activation leads to increased phospholipase C activity. To date, only inactivating mutations that cause endocrine dysfunction have been reported for the TRH receptor. One patient was identified with central hypothyroidism caused by mutated TRH receptors. He presented during the ninth year of life with short stature (− 2.6 SD), accompanied by a delayed bone age (− 4.1 SD), a low plasma thyroxine level, and a normal plasma TSH level. Exogenous TRH did not induce an increase in plasma TSH and prolactin levels. He was found to be compound heterozygous for TRH receptor gene mutations, resulting in receptors that failed to bind TRH or induce IP3 production. Another family was identified with complete resistance to TRH because of a nonsense mutation in the TRHR (p.R17X) producing a TRH receptor that lacked the entire transmembrane domain. The proband was homozygous and presented with short stature, growth failure, and fatigue at age 11 years. He had a low free T 4 , with a low-normal TSH. TRH stimulation testing failed to stimulate TSH or prolactin. Surprisingly his 33-year-old sister who was also homozygous had escaped detection despite two normal pregnancies brought to term. She had no signs or symptoms of hypothyroidism but exhibited thyroid function tests similar to the proband. She breastfed normally. Both the proband and his sister had normal cognitive function. This report suggested that the TRH receptor is not essential for normal cognitive function or female fertility and lactation. The mouse model corroborates these findings.
Other Class A Receptors That Transduce Hormone Action
Free Fatty Acid Receptor 1
At the time a new GPCR is discovered, the ligand for the newly discovered receptor is often unknown. Thus until a specific ligand is discovered, these GPCRs are known as orphan receptors . According to the Human Genome Organization (HUGO) Gene Nomenclature Committee, these G protein–coupled orphan receptors should be named alphanumerically GPR followed by a number, until their ligand is known. Once a specific ligand is identified, a more specific name is given the receptor.
The ligands for GPR40 were unknown when the receptor was first discovered. The HUGO Gene Nomenclature Committee changed the name of the receptor to free fatty acid receptor 1 (FFAR1) when the ligands were identified as medium- and long-chain fatty acids. With rare exceptions that are clearly identified, this chapter follows HUGO Gene Nomenclature Committee recommendations (see www.gene.ucl.ac.uk/nomenclature/index.html for more information on receptor nomenclature).
FFAR1 is one of several GRCRs for lipid mediators. Lipid mediators are intercellular lipid messengers that include sphingosine 1-phosphate, sphingosylphosphorylcholine, dioleoyl phosphatidic acid, lysophosphatidic acid, eicosatetraenoic acid, bile acids, and free fatty acids. FFAR1 is activated by medium- and long-chain fatty acids, whereas FFAR2 (formerly known as GPR43 ) and FFAR3 (formerly known as GPR41 ) are activated by shorter-chain fatty acids. There is now evidence that FFAR1 activation by medium- and long-chain fatty acids has endocrine implications. FFAR1 is expressed in human pancreatic β-islet cells. FFAR1 is involved in cholecystokinin secretion from I cells in response to fatty acids. It has also been implicated in fatty acid stimulated GLP-1 and GIP secretion from L and K cells. GPR120 is expressed in enteroendocrine cells and has a physiologic role in GLP-1 secretion. FFAR2 and FFAR3 are expressed in adipose tissue and FFAR3 has been implicated in leptin production.
Fatty acid–induced stimulation of FFAR1 in β-islet cells leads to activation of the Gα q -phospholipase C second-messenger pathway, which in turn leads to release of calcium from the ER that augments insulin-mediated increases in intracellular calcium concentrations because of glucose-induced activation of voltage-gated calcium channels. Because an increased intracellular calcium concentration induces insulin release, FFAR1-mediated augmentation of glucose-mediated increases in the intracellular calcium concentration leads to amplification of glucose-stimulated insulin release.
A variant in FFAR1 (p.Gly180Ser), found in a Sicilian population, resulted in obesity, impaired glucose tolerance, and lipid stimulated insulin secretion. Two other variants, p.Arg211His and p.Asp175Asn, are not associated with alterations in insulin release. TAK-875, an FFAR1 agonist, was shown to reduce hemoglobin A1c in patients with type 2 diabetes, in a phase 2 clinical trial. Wild-type mice placed on an 8-week high-fat diet develop glucose intolerance, insulin resistance, hypertriglyceridemia, and hepatic steatosis—whereas FFAR1 knockout mice on the same diet do not develop these conditions. The clinical relevance for patients is not yet clear. However, an Arg211His polymorphism in the FFAR1 gene may explain some of the variation in insulin secretory capacity found in Japanese men: Arg/Arg homozygotes had lower serum insulin levels, homeostasis model of insulin resistance, and homeostasis model of beta-cell function than His/His homozygotes.
KISS1 Receptor/GPR54
Studies in animal models suggests that Kiss1-expressing neurons in the hypothalamus modulate GnRH expressing neurons to initiate puberty and modulate sex steroid feedback on GnRH release. Homozygous inactivating mutations in the gene encoding the KISS1 receptor (GPR54) were initially described in French and Saudi Arabian patients with IHH; in both cases the affected subjects came from consanguineous families. The Saudi patients carried a p.Leu148Ser mutation, whereas the French patients carried a 155bp deletion. Leu148 is highly conserved among class A GPCRs. The mutation does not affect expression, ligand binding, or association with G s , but impairs ligand-induced catalytic activation of G s . At the same time, an African American patient with IHH was described who was compound heterozygous for inactivating GPR54 mutations. Since publication of these initial reports, additional patients have been described. A boy with a Jamaican father and a Turkish-Cypriot mother, and with cryptorchidism and micropenis at birth, and undetectable LH and FSH levels at 2 months of age, was found to have compound heterozygous GPR54 mutations. Another missense mutation (p.Leu102Pro) that exhibits complete inactivation of GPR54 signaling has been identified. Surprisingly, patients with this mutation exhibited spontaneous pulsatile LH and FSH secretion with normal frequency, and a blunted amplitude and family members had partial pubertal development. Biallelic loss of function mutations in GPR54 are a rare cause of normosmic IHH. A study of genetic causes of normosmic congenital hypogonadotropic hypogonadism showed that KISS1R mutations accounted for 2% of the variants.
Unlike patients with KS, but similar to patients with GnRH mutations, patients with GPR54 mutations have an intact sense of smell. In contrast to patients with IHH caused by GnRH mutations, patients with GPR54 mutations increase serum gonadotropin levels in response to exogenous GnRH.
Ligands for GPR54 derive from a single precursor protein, kisspeptin-1. The longest derivative protein that acts as a ligand for GPR54 is metastin, so called because it is a metastasis suppressor gene in melanoma cells. Metastin consists of kisspeptin-1 69-121. However, shorter carboxy-terminal peptides derived from kisspeptin-1 bind and activate GPR54. Administration of metastin to adult male volunteers increases LH, FSH, and testosterone levels.
An activating mutation in GPR54 was identified in a patient with central precocious puberty. The adopted girl was found to have an Arg386Pro mutation, which led to prolonged activation of signaling in response to kisspeptin. Mutational analysis of 28 subjects with idiopathic central precocious puberty failed to find any variants in KISS1 or KISS1R. A larger study showed an association between several variants and central precocious puberty in Korean girls.
Orexin Receptors
Orexins act on specific receptors that are located predominantly in the hypothalamus to control food intake and play a role in the regulation of sleep/wakefulness. There are two types of orexin receptors: the orexin-1 and the orexin-2 receptors. There are also two types of orexins, orexin A and orexin B, formed from the precursor peptide preproorexin. Orexins are also known as hypocretins , and orexin A is synonymous with hypocretin-1 and orexin B with hypocretin-2. Orexin A acts on orexin-1 and orexin-2 receptors, whereas orexin B only acts on orexin-2 receptors.
Like most class A GPCRs, orexin receptors couple with G q/11 and G i /G o to activate phospholipase C and inhibit adenylyl cyclase, respectively. Surprisingly, evidence suggests that orexin receptors also couple with G s —which increases adenylyl cyclase activity. Orexins increase food intake and duration of wakefulness. Orexin A and activation of the orexin-1 receptor have greater orexigenic effects than orexin B and activation of the orexin-2 receptor. The orexin-2 receptor mediates the arousal effect of orexins. Most patients with narcolepsy with cataplexy have diminished levels of orexin A concentrations in cerebral spinal fluid and lack orexin-containing neurons. This is thought to be because of postnatal cell death of orexin neurons in the hypothalamus. HLA DQB1*0602 is associated with narcolepsy with cataplexy and an autoimmune process has been suggested, but no autoantibodies have been identified. Thus far, no mutations in orexin receptors have been found in humans. A mutation in the orexin-2 receptor causes narcolepsy in dogs. There is one described mutation (p.leu16ARG) in the HCRT gene in a child with early-onset narcolepsy with cataplexy. This mutation was shown to impair processing and trafficking of the mutant orexin, leading to undetectable orexin A concentrations in the cerebrospinal fluid.
Ghrelin Receptors
The ghrelin receptor is also known as the GH secretagogue receptor type 1a because activation of receptors in the hypothalamus and pituitary somatotrophs enhances GH secretion. Ghrelin is a product of posttranslational modification of the ghrelin gene product proghrelin. Ghrelin is mainly produced in the stomach. Activation of ghrelin receptors located in the hypothalamus induces growth hormone–releasing hormone (GHRH) secretion. At the level of pituitary somatotrophs, it stimulates GH secretion. The ghrelin receptor also stimulates the appetite (i.e., an orexigenic role).
Plasma ghrelin levels are elevated just before eating and decrease rapidly after eating. In addition, intravenous administration of ghrelin to humans increases appetite and food intake. Plasma ghrelin levels are elevated in individuals with Prader Willi syndrome. Thus hyperphagia in patients with Prader Willi syndrome may be caused at least in part by overactivation of ghrelin receptors by ghrelin. Screening of 184 extremely obese children and adolescents for mutations of the ghrelin receptor gene failed to identify a single mutation likely to cause obesity. In contrast, large-scale screening studies have identified mutations and single nucleotide polymorphisms (SNPs) scattered throughout the ghrelin receptor, and two SNPs and one mutation in the promoter region, associated with increased transcriptional activity, cosegregated with obesity.
Short individuals in two unrelated Moroccan kindreds were found to have a C to A transversion at position 611 in the first exon of the ghrelin receptor gene. This transversion results in replacement of the apolar and neutral amino acid alanine at position 204 of the receptor by the polar and charged amino acid glutamate (p.ala204glu). This mutation interferes with normal constitutive activity of the receptor and decreases cell membrane expression of the receptor. Receptor activation by ghrelin, however, is preserved. Two-thirds of heterozygous individuals in the kindreds studied had short stature, with height more than or equal to 2 SDs below the mean. One heterozygous individual’s height was –3.7 SDs below the mean. Before onset of GH therapy, the only individual in the kindreds homozygous for the mutation had a height –3.7 SD, and became overweight during puberty. The weight of the patients heterozygous for the mutation varied from underweight to overweight. Another patient who presented with severe short stature (− 3 SD), vomiting, ketosis, hypoglycemia, and low BMI was identified to be a compound heterozygote for a p.W2X and an p.R237W mutation in the ghrelin receptor. His serum insulin-like growth factor-1 (IGF-1) level was low at 44 ng/mL and he failed GH stimulation testing, but had a normal IGF-1 generation test. This patient had improvement in his height velocity and resolution of hypoglycemia after treatment with GH. Four more ghrelin receptor mutations were identified in a Japanese cohort with short stature (p.Q36del, p.P108L, p.C173R, and p.D246A). p.Q36del showed a minor reduction in activity. C173R led to intracellular retention. D246 caused impaired signaling, and P108L led to reduced binding affinity to ghrelin. Two other mutations were identified in a cohort of patients with constitutional delay of growth and puberty in Brazil (Ser84Ile and Val182Ala). Both resulted in decreased basal activity. The patients were short at presentation (− 2.4 and − 2.3 SD) but reached a normal adult height without treatment.
Another product of posttranslational modification of proghrelin, obestatin, appears to play a role in controlling appetite and weight. Activation of the obestatin receptor, previously known as GPR39 , in rats results in decreased food intake and weight.
Melanin-Concentrating Hormone Receptors
Formerly known as SLC-1 or GPR24 , the type 1 melanin-concentrating hormone (MCH) receptor (MCHR1)—and the more recently discovered type 2 MCH receptor (MCHR2), formerly known as SLT or GPR145 —may play a role in regulating feeding and energy metabolism in humans. When activated, MCHR1 couples with G q/11 and G i/o to increase phospholipase C activity and inhibit adenylyl cyclase activity, respectively. MCHR2 couples with G q/11 , and MCH binding leads to increased phospholipase C activity.
Studies in rodents reveal that MCH is an orexigenic hormone, and treatment of rodents with MCHR1 antagonists decreases food intake, weight, and body fat. MCHR2 is not expressed in rodents. Deletion of the MCHR1 in mice prevents overeating in response to food cues under sated conditions. Two loss-of-function mutations were identified in MCHR1 in humans (R210H and P377S). Cells transfected with either mutant receptor failed to respond to MCH, despite normal cell-surface expression of the receptor, suggesting a receptor activation defect. These mutations were identified in two markedly underweight individuals and were not found in an obese cohort. Analysis of the MCHR1 gene in more than 4000 obese German, Danish, French, and American children and adolescents revealed several SNPs and gene variations in the German children and adolescents that may be associated with obesity. Another study of 106 American subjects with early onset obesity failed to definitively identify MCHR1 and MCHR2 mutations as a cause of obesity.
Class B receptors that transduce hormone action
Growth Hormone–Releasing Hormone Receptor
The GHRH receptor gene is located at 7p14. GHRH receptors interact with G s to stimulate adenylyl cyclase, resulting in increased intracellular cyclic AMP levels that lead to somatotroph proliferation and GH secretion. Thus it is not surprising that activating mutations in Gα s leading to constitutive activation of adenylyl cyclase have been found in some GH–secreting pituitary adenomas in humans.
Many mutations in the GHRH receptor that cause isolated GH deficiency have been identified. These include six splice site mutations, two microdeletions, two nonsense mutations, one frameshift mutation, 10 missense mutations, and one mutation in the promoter. The first naturally occurring mutation in the GHRH receptor (p.D60G) was found in the little mouse, which has a dwarf phenotype. This mutation in a conserved amino acid in the extracellular domain impairs the ability to bind GHRH. The first human mutation in the GHRH receptor (p.Glu72X) was identified in a consanguineous Indian family. The same mutation was found in three apparently unrelated consanguineous kindreds from India, Pakistan, and Sri Lanka. A different mutation (5’ splice site mutation in intron 1) was identified in a large Brazilian kindred of more than 100 individuals. Both mutations result in the production of markedly truncated proteins with no receptor activity. A frameshift mutation was identified in a patient with severe short stature and was the first documented case of early-onset anterior pituitary hypoplasia. In another family, two siblings with isolated GH deficiency were found to be compound heterozygous for inactivating GHRH receptor gene mutations. Three more novel mutations were identified in families with severe short stature in the United Kingdom (p.W273S, p.R94L, and p.R162W). The only mutation found in the promoter region of GHRH affects one of the Pit-1 binding sites.
Studies of subjects in these large kindreds who are homozygous or compound heterozygous for inactivating GHRH receptor gene mutations have shown that affected children experience severe postnatal growth failure with proportionate short stature. Males have high-pitched voices and moderately delayed puberty. Unlike infants with complete GH deficiency, they do not have frontal bossing, microphallus, or hypoglycemia. Other features include a doll facies, reduced muscle mass, central adiposity, wrinkled thin skin, and delayed pigmentation of the hair in children and teens. Bone age is delayed with respect to chronologic age, but advanced with respect to height age. Some patients have been found to have pituitary hypoplasia. Growth velocity increased with exogenous GH therapy. Remarkably, GH treatment of two siblings from Turkey, with the p.E72X mutation, allowed them to reach a normal adult height, despite pretreatment heights of − 6.7 and –8.6 SD and initiation of GH around age 14 years.
Further studies in the Brazilian cohort revealed that homozygotes had increased abdominal obesity, a higher low-density lipoprotein (LDL), and total cholesterol but normal carotid wall thickness and no evidence of premature atherosclerosis. Treatment of these patients with GH for 6 months improved body composition, reduced LDL and total cholesterol, and increased high-density lipoprotein (HDL). Surprisingly, this was associated with increased carotid intima-media thickness and atherosclerotic plaques. Reevaluation, 5 years after the discontinuation of GH, showed a return to baseline for these measures. Patients with a null mutation affecting the GHRH receptor also exhibited altered sleep patterns with abnormalities in both rapid eye movement and nonrapid eye movement sleep. Heterozygotes for the null mutation had normal adult heights and IGF-1 SD scores but exhibited reduced body weight, BMI, lean mass, fat mass, and increased insulin sensitivity.
SNPs in the GHRH receptor have been shown to contribute to height-SDS variation, but mutations remain a rare cause of isolated GH deficiency.
Gastric Inhibitory Polypeptide Receptors
The gastric inhibitory peptide receptor ( GIPR ) gene is located on the long arm of chromosome 19. Two functional isoforms exist in humans because of alternate splicing. GIPR activation induces Gα s activation of adenylyl cyclase. Gastric inhibitory polypeptide (GIP) is also known as glucose-dependent insulinotropic polypeptide and is released by K cells in the small intestine in response to food. GIP has numerous physiologic actions, including stimulation of glucagon, somatostatin, and insulin release by pancreatic islet cells. Human mutations in the GIPR have not been identified to date. One study identified an SNP in the GIPR that is associated with insulin resistance in obese German children. Another study identified an SNP in the GIPR that is associated with reduced fasting and induced C-peptide levels.
The GIPR is implicated in food-dependent Cushing syndrome. Circulating cortisol levels in patients with food-dependent or GIP-dependent Cushing syndrome rise abnormally in response to food intake. GIP does not normally induce cortisol release from adrenocortical cells. These patients may have adrenal adenomas or nodular bilateral adrenal hyperplasia that overexpresses GIPRs that abnormally stimulate cortisol secretion when activated. Thus in these patients postprandial GIP release leads to activation of these abnormally expressed and functioning adrenal GIPRs, resulting in excessive adrenal cortisol secretion.
Parathyroid Hormone and Parathyroid Hormone–Related Peptide Receptors
Two types of PTH receptors have been identified. The type 1 PTH receptor (PTHR1) is activated by PTH and parathyroid hormone–related peptide (PTHrP) and mediates PTH effects in bone and kidney. In spite of 51% homology to the PTHR1, the type 2 PTH receptor (PTHR2) is activated by PTH but not PTHrP. The PTHR2 is particularly abundant in the brain and pancreas but is also expressed in the growth plate; its natural ligand is TIP39. The function of the PTHR2 is largely unknown.
The PTHR1 has a large amino-terminal extracellular domain containing six conserved cysteine residues. Ligand binding induces the PTHR1 to interact with G s and G q proteins, leading to activation of the adenylyl cyclase/PKA and phospholipase C/protein kinase C second-messenger pathways, respectively. Interestingly, mutations in i2 interfere with coupling of the PTHR1 to G q , without interfering with coupling to G s —whereas mutations in i3 disrupt coupling of the receptor to both G proteins. Binding of PTH to the PTHR1 leads to internalization of a portion of plasma membrane containing a ternary complex of activated receptor-Gsα-adenylyl cyclase that exhibits sustained production of cyclic AMP. In contrast, PTHrP binding to the PTHR1 receptor leads to formation of a complex that remains on the cell surface and generates cyclic AMP for only a short period of time.
Biallelic loss-of-function mutations in the PTHR1 gene cause Blomstrand chondrodysplasia. This lethal disorder is characterized by accelerated chondrocyte differentiation, resulting in short-limbed dwarfism, mandibular hypoplasia, lack of breast and nipple development, and severely impacted teeth. One patient with this rare condition was found to be homozygous for a point mutation that resulted in a p.Pro132Leu substitution in the amino-terminal domain that interferes with ligand binding. Another patient was found to be homozygous for a frameshift mutation that results in a truncated receptor lacking TM5-7, and contiguous intracellular and extracellular domains.
A third patient was found to have a maternally inherited mutation that altered splicing of maternal mRNA, resulting in a PTHR1 with a deletion of residues 373 through 383 in TM5 (which also interferes with ligand binding). In spite of heterozygosity for the mutation, the patient was unable to produce normal PTHR1s, because for unknown reasons the paternal allele was not expressed. Heterozygosity for somatic or germline p.Arg150Cys missense mutations in the PTHR1 was identified in two out of six patients with enchondromatosis, a condition that is usually sporadic and which is attributed to a postzygotic somatic cell mutation. Enchondromas are benign cartilage tumors that develop in the metaphyses and may become incorporated into the diaphyses of long tubular bones, in close proximity to growth plate cartilage; there is an increased risk of malignant transformation to osteosarcoma. Patients with multiple enchondromatosis (OMIM ID: 166000) have Ollier disease (World Health Organization terminology), a disorder characterized by the presence multiple enchondromas with an asymmetric distribution of lesions that vary in size, number, and location. When multiple enchondromatosis occurs with soft tissue hemangiomas, the disorder is known as Maffucci syndrome . In vitro studies showed that the p.Arg150Cys mutation was mildly activating but led to stimulation of phospholipase C rather than adenylyl cyclase. A transgenic knockin mouse expressing the mutant PTHR1, under the control of the collagen type 2 promoter, showed development of tumors that are similar to those observed in human enchondromatosis. The clinical significance of these observations are uncertain, as most cases of enchondromatosis are caused by mutations in the IDH1 and IDH2 genes.
Biallelic loss of function mutations in PTHR1 are also the cause for Eiken syndrome, which is characterized by a skeletal dysplasia with severely retarded ossification, principally of the epiphyses, pelvis, hands, and feet, as well as abnormal modeling of the bones. Duchatelet et al. mapped Eiken syndrome to chromosome 3p near the PTHR1 gene. Affected individuals were homozygous for a nonsense mutation in the carboxy-terminal cytoplasmic tail of the PTHR1 gene (p.R485X). In a 7-year-old boy with Eiken syndrome, who was born to unaffected first-cousin parents, Moirangthem et al. identified a homozygous missense mutation at a conserved residue in the PTHR1 gene (p.E35K). Finally, nonsyndromic primary failure of tooth eruption (PFE) is caused by heterozygous mutations in the PTHR1 gene. Three distinct mutations, namely c.1050-3C > G, c.543 + 1G > A, and c.463G > T, were identified in 15 affected individuals from four multiplex pedigrees. All mutations truncate the mature protein and therefore should lead to a functionless receptor.
Some cases of Jansen metaphyseal chondrodysplasia have been found to be caused by constitutively activating mutations of the PTHR1 gene. This autosomal-dominant disorder is characterized by short-limbed dwarfism caused by impaired terminal chondrocyte differentiation and delayed mineralization, accompanied by hypercalcemia. Interestingly, constitutive activation appears to result predominantly in excessive Gα s activity because adenylyl cyclase activity is increased and phospholipase C activity is unchanged in COS-7 cells expressing mutated receptors.
Other Class B Receptors That Transduce Hormone Action
Other class B receptors that transduce hormone action include glucagon-like peptide-1, glucagon, calcitonin, and corticotrophin-releasing factor receptors. Class B receptors usually couple with heterotrimeric G s proteins, leading to the activation of adenylyl cyclase—which in turn leads to elevated intracellular cyclic AMP levels. (See chapter on Diabetes Mellitus. for a discussion of the role of GLP1 in promoting insulin secretion and the use of GLP1 analogues or inhibitors of GLP1 breakdown in therapy.)
Class C receptors that transduce hormone action
Calcium-Sensing Receptors
The calcium-sensing receptor (CaSR) is located on the long arm of chromosome 3 (3q21.1). The CaSR has a large amino-terminal domain that contains nine potential glycosylation sites. Binding of ionized calcium to the CaSR leads to activation of phospholipase C via activation of G q/11 proteins.
The CaSR is an integral component of a feedback system that uses PTH and renal tubular calcium reabsorption to keep the serum concentrations of ionized calcium within a narrow physiologic range. Increased extracellular ionized calcium concentrations activate CaSRs in parathyroid chief and renal tubular epithelial cells, leading to decreased PTH release and renal tubular calcium reabsorption. When ionized calcium concentrations fall, CaSR activation decreases—leading to increased PTH release and enhanced renal tubular calcium reabsorption.
The CaSR also binds magnesium, and thus PTH secretion can be inhibited by elevated serum concentrations of magnesium with consequent hypocalcemia. The CaSR may participate in magnesium homeostasis by altering reabsorption of magnesium in the thick ascending limb of Henle in the kidneys. It is probable that increased peritubular levels of magnesium activate renal CaSRs, leading to inhibition of reabsorption of magnesium from the thick ascending limb of Henle—which in turn leads to increased renal excretion of magnesium.
Both loss of function and gain of function mutations in the CaSR have been described in patients with hypocalcemia and hypercalcemia, respectively. Familial (benign) hypocalciuric hypercalcemia type 1(FHH1) and neonatal severe hyperparathyroidism (NSHPT) are caused by loss-of-function mutations of the CaSR gene. Most of these mutations are located in the amino-terminal extracellular domain. With few exceptions, individuals heterozygous for loss-of-function mutations have FHH1, a benign condition characterized by very low urinary calcium excretion, mild hypercalcemia, normal or slightly elevated serum PTH levels, and few if any symptoms of hypercalcemia or hyperparathyroidism. In contrast, individuals homozygous for such mutations will develop NSHPT, a life-threatening condition characterized by severe hypercalcemia, markedly elevated serum PTH levels and skeletal defects. Therefore children of consanguineous FHH parents are at risk for NSHPT. Occasionally, infants with NSHPT are heterozygous for CaSR gene mutations that encode a dominant negative receptor protein. In most cases, FHH1 is transmitted in an autosomal dominant manner, but autosomal recessive inheritance has been described in one kindred in which the CaSR mutation was only weakly inactivating.
Decreased CaSR function impedes calcium ion suppression of PTH release and renal tubular calcium reabsorption. Thus FHH1 is characterized by mild hypercalcemia that is accompanied by inappropriately normal or elevated serum PTH levels and by relatively low urinary calcium excretion. Individuals with FHH1 may also have hypermagnesemia as a result of decreased peritubular inhibition of magnesium reabsorption from the thick ascending loops of the kidneys by the CaSR. In addition to FHH1, there are two other variants, FBH type 2 (FBH2) and FBH type 3 (FBH3), that are caused by heterozygous loss of function mutations in GNA11 and AP2S1, respectively.
FHH1 is caused by heterozygous loss-of-function mutations of the CaSR gene on 3q21.1. Two other chromosomal loci have been identified in patients with FBH who do not have CaSR gene mutations. FHH2 has been mapped to GNA11 , which encodes the α subunit of G11, located 19p13.3 and is biochemically and clinically similar to FHH1. FBH3, which is also known as the Oklahoma variant (FHH OK ), is caused by mutations in the AP2S1 gene located at 19q13. Adults with FHH3 have hypophosphatemia, elevated serum PTH levels, and osteomalacia, in addition to the clinical and biochemical findings found in individuals with FHH1 and FHH2. NSHPT is characterized by severe hypercalcemia accompanied by elevated circulating PTH levels, undermineralization of bone, rib cage deformity, and multiple long-bone and rib fractures.
Activating mutations of the CaSR gene cause autosomal-dominant hypocalcemia type 1 (ADH1), as increased CaSR function leads to increased calcium ion suppression of PTH release and suppression of renal tubular calcium reabsorption. ADH1 is characterized by hypocalcemia and hypomagnesemia, accompanied by inappropriately normal or increased urinary calcium excretion and inappropriately normal or low serum PTH levels. Patients with ADH1 may be asymptomatic or may present with tetany, muscle cramps, or seizures during infancy or childhood. Similar to inactivating mutations, most activating mutations are located in the amino-terminal extracellular domain. Treatment of patients with ADH1 with activated forms of vitamin D (e.g., calcitriol) is complicated, as normalization of serum calcium levels is associated with worsening of hypercalciuria, hence further increasing the risk of nephrolithiasis, in nephrocalcinosis, and in renal impairment. In contrast, urinary calcium excretion is not excessive in patients with ADH2, who have activating mutations in GNA11 . GNA11 encodes the α subunit of G11, the principle G protein that couples to CaSR in parathyroid cells but not in the kidney.
Some patients with activating mutations of the CaSR gene will develop Bartter syndrome type V, which similar to other types of Bartter syndrome, is characterized by hypokalemic metabolic alkalosis and by hyperaldosteronism caused by elevated renin levels. Patients with Bartter syndrome type V, unlike patients with other types of Bartter syndrome, may also have symptomatic hypocalcemia and are at risk for developing nephrocalcinosis because of hypercalciuria. Evidence from in vitro functional expression studies suggests that patients with mild or moderate heterozygous gain-of-function mutations of the CaSR develop ADH1, whereas those with severe heterozygous gain-of-function mutations of the CaSR will also develop Bartter syndrome type V.
Some single–amino-acid polymorphisms of the CaSR gene appear to predict whole-blood ionized and serum total calcium levels and may increase the risk for bone and mineral metabolism disorders in individuals with other genetic and environmental risk factors for these disorders. Individuals heterozygous or homozygous for a Gln1011Glu CaSR gene polymorphism tend to have higher calcium levels than individuals with the polymorphism. The 15.4% of 387 healthy young Canadian women, with at least one CaSR gene allele with an Ala986Ser polymorphism, were found to have higher total calcium levels than the remainder of the women without the polymorphism.
Another study of 377 unrelated healthy Italian adult males and females found that 24% of study subjects were heterozygous or homozygous for the p.Ala986Ser polymorphism and confirmed the finding that individuals without the polymorphism have lower whole-blood ionized calcium levels than individuals with the polymorphism. The p.Ala986Ser polymorphism has also been associated with Paget disease and primary hyperparathyroidism. Individuals with a less common Arg990Gly polymorphism tend to have lower whole-blood ionized calcium levels than individuals without the polymorphism. The p.Arg990Gly polymorphism has been found to be associated with hypercalciuria and nephrolithiasis.
Autoantibodies against the CaSR that interfere with binding of calcium to the receptor may cause autoimmune hypocalciuric hypercalcemia. These patients have primary hyperparathyroidism with the clinical and biochemical features of patients with FHH1. Conversely, autoantibodies that activate the CaSR are a cause of autoimmune-acquired hypoparathyroidism. Both conditions may occur in association with other autoimmune conditions (such as autoimmune thyroiditis), with celiac disease in patients with autoimmune hypocalciuric hypercalcemia, and with autoimmune thyroiditis and autoimmune polyglandular syndrome types 1 and 2 in patients with autoimmune-acquired hypoparathyroidism. Autoantibodies that activate the CaSR were found in approximately one-third of individuals with acquired hypoparathyroidism.
G protein gene disorders
A growing number of human disorders are associated with somatic or germline mutations in genes that encode the subunits of G proteins and lead to either a gain of function or a loss of function in the signaling protein. Here, we will limit discussion to disorders associated with mutations of the GNAS gene that encodes Gα s , as this is the most common G protein to cause endocrine disorders.
Inactivating Mutations of the GNAS Gene
Pseudohypoparathyroidism type 1a (PHP1a; Albright hereditary osteodystrophy [AHO]) and pseudopseudohypoparathyroidism (PPHP) are caused by heterozygous inactivating mutations of the GNAS gene that encodes Gα s . PHP1a is characterized by end organ resistance to PTH with consequent hypocalcemia, hyperphosphatemia, and elevated circulating PTH levels. In addition, patients also manifest resistance to other hormones whose receptors couple to Gs, such as GHRH, TSH, gonadotropin, calcitonin, and hypothalamic neurotransmitters. PHP1a patients also have neurocognitive impairment and obesity that reflect the effect of the Gα s in the brain. In addition, patients manifest a constellation of developmental defects that have been termed Albright hereditary osteodystrophy and which include heterotopic ossifications, short stature, craniofacial anomalies, and brachydactyly D/E of the hands and feet, characterized by shortened fingers and short fourth and fifth metacarpals. Patients with the associated condition PPHP share the same features of AHO as PHP1a, but do not have hormone resistance. The distinction between these two manifestations of the same gene defect is not stochastic but results from a complex mechanism of genomic imprinting that controls transcription of the GNAS gene. Hence, patients with a GNAS mutation on a maternal allele will develop a more severe form of Gsα deficiency with hormone resistance (i.e., PHP1a), whereas patients with identical mutations on the paternal GNAS allele will have a milder condition with normal hormone responsiveness (i.e., PPHP).
Although most cells express Gsα from both parental alleles, in some cells Gsα expression is suppressed from the paternal GNAS allele. Thus patients with PHP1a develop hormone resistance that is limited to the thyroid, pituitary somatotrophs, and proximal renal tubule cells because in these cells, Gα s is derived principally from the maternal GNAS allele. Thus patients with maternally inherited inactivating GNAS gene mutations express very little Gα s in these cells and develop hormone resistance. Because Gsα is not expressed from the paternal allele in imprinted tissues, PPHP patients with paternally inherited inactivating GNAS gene mutations do not experience a deficit in Gsα protein in these cells, as they will have a normal amount of Gsα protein that is produced from the wild-type maternal GNAS allele. Patients with either PHP1a or PPHP express only 50% of the normal amount of Gsα protein in cells in which Gsα transcription is not controlled by the imprinting mechanism, which leads to haploinsufficiency, and likely accounts for the similar features of AHO that occur in these two conditions. Subjects with paternally inherited GNAS mutations have variable features of AHO without hormonal resistance and have been described to have PPHP, progressive osseous heteroplasia (POH), or osteoma cutis, based on the clinical phenotype. The basis for these distinctions is unknown.
Subjects with PHP type 1b (PHP1b; MIM 603233) lack typical features of AHO, but may have mild brachydactyly. PTH resistance is the principal manifestation of hormone resistance, but some patients have slightly elevated serum levels of TSH and normal serum concentrations of thyroid hormones as evidence of partial TSH resistance. It was initially thought that PHP1b is caused by inactivating mutations in the PTHR1 gene. However, no deleterious PTHR1 gene mutations have been found in patients with PHP1b.
Epigenetic defects in imprinting of GNAS are the cause of PHP1b. There are three alternative first exons for GNAS (i.e., NESP55, XLαs, and exon A/B) that splice onto exons 2 through 13 and are associated with differentially methylated regions (DMRs). Most relevantly, the exon A/B DMR is methylated in the maternal germ cells and seems to be the principal control element for transcription from exon 1. Loss of methylation in the DMR upstream of exon A/B of the maternal allele is a consistent finding in patients with PHP1b, and this epigenetic defect accounts for decreased Gsα expression from the affected allele. Most cases of autosomal dominant PHP1b are caused by microdeletions on the maternal allele that include exons 3 to 5 or 4 to 6 of the gene encoding syntaxin-16 ( STX16 ). Three other maternally inherited microdeletions involving NESP55 and AS (delNESP55/ASdel3-4) or AS (delAS3-4) alone have been identified, and these deletions produce a more global disruption of methylation that includes three GNAS DMRs (A/B, XL/AS, and NESP55). The genetic basis for most cases of sporadic PHP1b remains unknown and does not appear to be associated with the GNAS locus. These patients have global epigenetic defects in methylation that affect all three DMRs. In some cases, partial or complete paternal uniparental disomy (UPD) for chromosome 20 has been identified, in which two normal copies of GNAS are both derived from the father. Paternal UPD would predict a near complete deficiency of Gα s in imprinted cells and tissues in which Gα s is not transcribed from the paternal allele.
Gsα is not expressed in the renal proximal tubules of these patients because of lack of a GNAS allele with a maternal epigenotype. In contrast, both of the GNAS alleles with paternal epigenotypes are normally expressed in nonimprinted cells in which expression of Gsα from the paternal allele is not suppressed.
Activating Mutations of the GNAS Gene
When a GPCR is activated by a ligand, the activated receptor interacts with the heterotrimeric G protein and allows release of bound GDP from the Gα subunit with replacement by GTP. The GTP-bound Gα dissociates from the Gβγ dimer, and both complexes are free to associate with downstream signal generating molecules. The generation of second messengers is terminated by an intrinsic GTPase within the Gα subunit that acts as a timer; hydrolysis of GTP to GDP inactivates Gα and increases its affinity for Gβγ—leading to reassociation of an inactive heterotrimeric G protein that is ready for another cycle of receptor-induced activation. For Gsα, the amino acids Arg201 and Gln227 are critical to GTPase activity. GNAS mutations that result in substitutions of these amino acid residues lead to abrogation of GTPase activity and therefore prolong the active state of Gα s , thereby leading to constitutive (i.e., receptor ligand-independent) stimulation of adenylyl cyclase. Somatic mutations of these residues that disrupt GTPase activity are present in approximately 40% of GH–secreting and some ACTH-secreting and nonsecreting pituitary tumors; in some parathyroid, ovarian, testicular, thyroid, and adrenal tumors; and in some intramuscular myxomas.
More widespread mosaic Arg201 Gα s mutations that decrease GTPase activity cause fibrous dysplasia or (when tissue distribution of the mutation is very widespread) McCune-Albright syndrome, which is characterized by the triad of café-au-lait spots, polyostotic fibrous dysplasia, and primary endocrine hyperfunction, particularly gonadotropin-independent precocious puberty.
Patients with McCune-Albright syndrome may also have excessive GH production, hyperthyroidism, and hypercortisolism, as well as nodularity of the pituitary, thyroid, and adrenal glands because of the growth-promoting effects of excessive cyclic AMP in these tissues. Hypophosphatemia, which is not uncommon in patients with McCune-Albright syndrome, appears to be caused by excessive production of the phosphatonin fiberblast growth factor (FGF)-23 by fibrous dysplasia skeletal lesions. Patients with McCune-Albright syndrome may also have nonendocrine problems, such as hepatobiliary abnormalities, cardiomyopathy, optic neuropathy, and sudden death.
Cytokine receptors
Cytokines are molecules produced by one cell that act on another cell. Thus the term cytokine can apply not only to molecules with immunologic functions, but also to hormones. Therefore GH, prolactin, and leptin are classified as type 1 cytokines. These and other type 1 cytokines (including interleukins [ILs] 2–9, 11–13, and 15; erythropoietin; thrombopoietin; and granulocyte-colony–stimulating factor) are characterized by a four α-helical bundle structure and signaling via type 1 cytokine receptors. Type 2 cytokines include the interferons and IL-10 and do not include hormones.
Type 1 cytokines are divided into long-chain and short-chain cytokines. Prolactin, leptin, and GH belong to the long-chain subclass of type 1 cytokines because their helixes are 25 amino acids in length. The short-chain type 1 cytokines, including IL-2 and stem cell factor, have helixes of approximately 15 amino acids in length.
Structure and Function of Type 1 Cytokine Receptors
All type 1 cytokine receptors have four conserved cysteine residues, fibronectin type 2 modules, a Trp-Ser-X-Trp-Ser motif in the extracellular domain, and a proline-rich Box 1/Box 2 region in the cytoplasmic domain. With the exception of stem cell factor, type 1 cytokine receptors do not contain catalytic domains, such as kinases.
Type 1 cytokine receptors for long-chain type 1 cytokines require homodimerization for activation. First, the ligand binds a monomeric receptor. Then, the ligand interacts with a second receptor to induce receptor dimerization and activation. Activated receptors then stimulate members of the Janus family of tyrosine kinases (Jak kinases) to phosphorylate tyrosine residues on both the kinase itself and the cytoplasmic region of the receptors. Signal transducers and activators of transcription (STATs) then dock on the phosphorylated cytoplasmic receptor domains or Jak kinases via an SH2 domain, and are tyrosine phophorylated. The phosphorylated STATs then dissociate from the receptors or Jak kinases, form homodimers or heterodimers, and translocate to the nucleus. In the nucleus, the STAT dimers bind and alter the activity of regulatory regions of target DNA.
There are four Jak kinases. Jak3 is only expressed in lymphohematopoietic cells, whereas Jak1, Jak2, and Tyk2 are expressed in every cell. There are seven STATs (Stat1, Stat2, Stat3, Stat4, Stat5a, Stat5b, and Stat6), which have different SH2 domain sequences that confer different receptor specificities.
Cytokine Receptors That Transduce Hormone Action
The actions of GH, prolactin, and leptin are mediated via specific type 1 cytokine receptors. Mutations of the growth hormone receptor (GHR) and the leptin receptor have been identified as the bases of specific endocrine disorders ( Table 3.3 ).
| Receptor | Germline Mutation | Endocrine Disorder |
|---|---|---|
| Growth hormone receptor | Some inactivating(heterozygous) Inactivating (homozygous, compound heterozygous) | Partial growth hormone insensitivity with mild to moderate growth failure Growth hormone insensitivity/Laron syndrome with moderate to severe postnatal growth failure |
| Leptin receptor | Inactivating (homozygous) | Obesity and hypogonadotropic hypogonadism |
Growth Hormone Receptors
The GHR gene is located on the short arm of chromosome 5 (5p13.1-p12), and 9 of the 13 exons of the gene encode the receptor. A secretion signal sequence is encoded by exon 2, the amino-terminal extracellular ligand binding domain is encoded by exons 3 through 7, the single transmembrane domain is encoded by exon 9, and the carboxy-terminal cytoplasmic domain is encoded by exons 9 and 10. Growth hormone binding protein (GHBP) is produced by proteolytic cleavage of the extracellular domain of the GHR from the rest of the receptor. Approximately 50% of circulating GH is bound to GHBP.
Binding of GH to its receptor induces receptor dimerization and association with JAK2, a member of the Janus kinase family, which results in self-phosphorylation of JAK2 and a cascade of phosphorylation of cellular proteins, including Stat1, Stat3, and Stat5. The most critical of these proteins is STAT5b, which couples GH binding to the activation of gene expression that leads to the intracellular effects of GH, including synthesis of IGF-1, insulin-like growth factor binding protein 3 (IGFBP3), and acid labile subunit (ALS). The phosphorylated STATs translocate to the nucleus, where they regulate GH–responsive genes. In particular, GH indirectly controls growth by regulating production of IGF-1—which has direct effects on cell proliferation and hypertrophy. Jak2 also activates the mitogen activated protein (MAP) kinase and insulin receptor substrate pathways. However, the extent to which these pathways contribute to GH action is as yet unknown.
Patients are considered to have growth hormone insensitivity (GHI) if they do not exhibit appropriate growth and metabolic responses to physiologic levels of GH. The phenotype of GHI is variable and ranges from isolated moderate postnatal growth failure to severe postnatal growth failure, accompanied by the classic features of Laron syndrome in Ecuadorean patients with GHR deficiency. Features of GHR deficiency include frontal temporal hairline recession, prominent forehead, decreased vertical dimension of face, hypoplastic nasal bridge, shallow orbits, blue sclera, small phallus before puberty, crowded permanent teeth, absent third molars, small hands and feet, hypoplastic fingernails, hypomuscularity, delayed age of onset for walking, high-pitched voice, increased total and LDL cholesterol, and fasting hypoglycemia. All patients with GHI have normal or elevated circulating GH levels, markedly decreased circulating IGF-1 levels, and a delayed bone age.
Patients homozygous or compound heterozygous for deletion of exons 5 and 6—or homozygous or compound heterozygous for numerous nonsense, missense, frameshift, and splice-point mutations throughout the GHR gene—have been found to have GHI characterized by severe postnatal growth failure and usually low or absent circulating GHBP levels. Patients homozygous or compound heterozygous for the p.Arg274Thr or the p.Gly223Gly splice mutations that result in a truncated receptor that cannot be anchored to the plasma membrane (or that result in the p.Asp152His missense mutation that interferes with GHR dimerization) have normal circulating GHBP levels.
Patients heterozygous for mutations that alter the GHR have dimerization complexes that consist of two wild-type receptors, a wild-type receptor and a mutant receptor, and two mutant receptors. Thus heterozygosity for loss-of-function GHR gene mutations may have a dominant negative effect because the wild-type receptor/mutant receptor dimers may not be able to function normally. As expected from this phenomenon, some patients with moderate to severe growth failure have been found to be heterozygous for loss-of-function point or splice mutations of the GHR gene that alter the cytoplasmic or extracellular domains. In some cases, nonsense-mediated mRNA decay can lead to degradation of the aberrant mRNA and prevent a potential dominant negative effect.
Some patients with severe short stature and GHI do not have GHR mutations. Rather, they have defects in GHR-mediated intracellular signaling—including impaired STAT activation. Some patients with GHI were found to have STAT5b mutations. Unlike patients with GHR mutations, patients with STAT5b mutations also exhibited severe neurocognitive delay, chronic lung disease (lung fibrosis and/or lymphoid, interstitial pneumonia, severe eczema, T-cell lymphopenia, and abnormal T-cell function. Patients with GHI can now be successfully treated with recombinant IGF-1, but this does not result in a normal adult height, in contrast to GH deficiency in which GH therapy can achieve normal adult height.
Leptin receptors
The leptin receptor ( LEPR , also known as Ob-R ) gene is located at 1p31. There are five isoforms of LEPR because of alternative splicing of the LEPR gene transcript ( Fig. 3.5 ). Only the Ob-Rb isoform contains both the Jak kinase binding and STAT motifs necessary to maximally transduce the effects of leptin. The Ob-Ra, Ob-Rc, and Ob-Rd isoforms contain intact extracellular and transmembrane but are missing the STAT motif from their cytoplasmic domains. The Ob-Re isoform is missing the transmembrane and cytoplasmic domains. Thus the Ob-Rb isoform is thought to be the main isoform involved in mediating the effects of leptin.
Three sisters from a consanguineous kindred were found to be homozygous for a splice mutation in the LEPR gene that resulted in expression of an 831-amino-acid protein (Ob-Rhd) that lacks transmembrane and cytoplasmic domains. They had been hyperphagic and morbidly obese since birth. They were found to have elevated circulating leptin levels, decreased TSH and GH secretion, and failure of pubertal development caused by hypogonadotropic hypogonadism. Heterozygous carriers of the mutation are not morbidly obese and do not have delayed or absent puberty.
Nonsense or missense LEPR mutations were identified in 3% of a selected cohort of 300 subjects with early-onset obesity. Individuals with mutations had hyperphagia, severe obesity beginning in the first year of life with a mean BMISDS of + 5.1, high rate of childhood infections, altered immune function, with moderately reduced CD4 counts (988 vs. 1100 cells/mL 3 ), and delayed puberty because of hypogonadotropic hypogonadism. Childhood growth was normal but final height was at –1.7 to –2 SD because of the lack of a pubertal growth spurt. Importantly, circulating leptin levels were within the range predicted by the elevated fat mass, and clinical features were less severe than those of subjects with congenital leptin deficiency. Heterozygous family members had an average BMISDS of + 0.6 similar to that of family members with normal leptin receptors. Functional characterization of these missense mutations revealed defects causing intracellular retention, misfolding, or failure to signal to downstream pathways. Additional novel mutations have been reported that cause similar phenotypes.
Receptor tyrosine kinases
The receptor tyrosine kinase (RTK) superfamily consists of 15 receptor tyrosine kinase families ( Fig. 3.6 ). With one exception, these families consist of receptors with one membrane-spanning domain (see Fig. 3.6 ). The single-membrane–spanning receptors typically contain an amino-terminal extracellular portion, a transmembrane helix, a juxtamembrane region, a tyrosine kinase (TK) domain, and a carboxy-terminal region (see Fig. 3.6 ). These receptors require dimerization to be maximally activated. Receptors belonging to the insulin RTK family differ from other RTKs, as they contain two membrane-spanning polypeptide chains, linked by disulfide bonds, to two intervening extracellular peptide chains and thus do not dimerize (see Fig. 3.6 ).
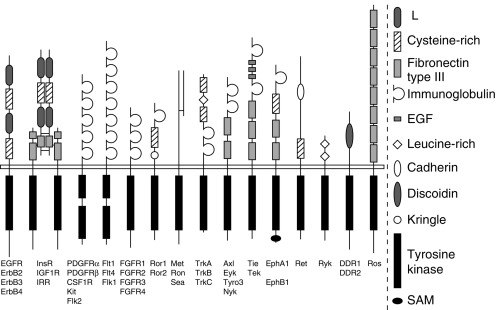
Activation of RTKs leads to phosphorylation of tyrosine residues in the activation loop (A-loop) in the TK domain(s), resulting in activation of the TK(s). Activation of the TK(s), in turn, induces the transfer of phosphate from adenosine triphosphate (ATP) to tyrosine residues in the cytosolic portion of the receptor and in cytosolic proteins that serve as docking sites for second messengers.
There is a growing body of evidence suggesting that members of the receptor TK superfamily can directly and indirectly interact with heterotrimeric G proteins. The insulin, IGF-1, and IGF-2 receptors appear to directly interact with G i/o and G q/11 , G i/o, and G i , respectively. The FGF receptors (FGFRs) appear to directly and indirectly interact with G s . Congenital alteration of function of receptors in the insulin and the FGF RTK families leads to endocrine disorders ( Table 3.4 ).
| Receptor | Germ-Line Mutation | Endocrine Disorder |
|---|---|---|
| Insulin receptor | Inactivating (heterozygous) | Some cases of type A syndrome |
| Inactivating (homozygous, compound heterozygous) | Rabson-Mendenhall, Donohue (leprechaunism), and some cases of type A syndromes | |
| IGF-1 receptor | Gene deletion (heterozygous) | Pre- and postnatal growth failure |
| FGFR1 | Inactivating mutation (heterozygous) | Kallmann syndrome, missing teeth, cleft palate |
| FGFR2 | Inactivating mutation (heterozygous) | Apert, Pfeiffer, Crouzon syndromes |
| FGFR3 | Activating mutations (heterozygous) | Achondroplasia, severe achondroplasia with developmental delay and acanthosis nigricans, thanatophoric dysplasia types I and II, and platyspondylic lethal skeletal dysplasias (San Diego types) |
Insulin receptor tyrosine kinase family
The insulin RTK family includes the insulin receptor (INSR) and the IFG-1 receptor (IGF1R). These receptors are heterotetramers consisting of two α and β subunits in an αββα configuration ( Fig. 3.7 ). The cysteine-rich extracellular α subunits are linked by disulfide bonds, and each α subunit is linked to a plasma membrane–spanning and cytosolic β subunit by disulfide bonds. Each β subunit contains a TK domain and a carboxy-terminal region that contain tyrosine residues.
Both insulin and IGF-1 can bind INSRs and IGF1Rs. However, insulin has greater affinity for the INSR and IGF-1 has greater affinity for the IGF1R. Ligand binding alters the conformation of the receptor, resulting in trans -autophosphorylation of the carboxy-terminal tyrosine residues on one β subunit by the TK on the other β subunit. The phosphorylated tyrosine residues create motifs that can be bound by Src homology 2 (SH2)-domain–containing proteins, including Shc, Grb-2, SHP2, nck, phosphatidylinositol-3-kinase (PI3K), and Crk. The receptor TK also phosphorylates tyrosine residues in insulin receptor substrate proteins (IRS), including IRS-1 and IRS-2, that bind INSRs and IGF1Rs. When phosphorylated, these tyrosine residues create motifs that are bound by SH2-domain–containing proteins. Thus insulin receptor substrates can serve as docking proteins—allowing SH2-domain-containing proteins to indirectly interact with INSRs and IGF1Rs, when stearic constraints do not permit direct interactions between the proteins and the receptors. Ultimately, IRSs, SH2-domain–containing proteins, and other proteins (including mSOS) interact to activate the Ras/Raf/MAPKK/MAPK and PI3K/protein kinase B (PKB) cascades (see Fig. 3.7 ).
Activation of the Ras/Raf/ MAPKK/MAPK cascade increases mitogenesis and proliferation, and activation of the PI3K/PKB cascade increases glucose uptake and glycogen synthesis. Evidence suggests that IGF-1 has a greater effect on cell growth than on glucose metabolism because activation of the IGF1R stimulates the Ras/MAPK cascade more than INSR activation. Conversely, it appears that insulin has a greater effect on glucose metabolism because INSR activation stimulates the PI3K/PKB cascade more than IGF1R activation.
The insulin receptor
The INSR gene is located on 19p and contains 22 exons. The αβ half-receptor precursors are derived from proteolysis of a single proreceptor comprised of α and β subunits in tandem and disulfide linkage of these subunits. These αβ half-receptor precursors then join to form a single αββα heterotetrameric insulin receptor. Interestingly, αβ half-receptor precursors encoded by one allele may combine with αβ half-receptor precursors encoded by the other allele to form a single insulin receptor. This phenomenon explains how heterozygous mutations resulting in impaired β subunit TK activity can have a dominant negative effect because activation of the INSR requires trans -autophosphorylation of one β subunit by the other β subunit.
Mutations in the insulin receptor lead to Donohue syndrome (leprechaunism), Rabson-Mendenhall syndrome, or type A insulin resistance syndrome. Patients with leprechaunism or Donohue syndrome are severely insulin resistant. They present during infancy with severe intrauterine and postnatal growth retardation, lipoatrophy, and acanthosis nigricans. They also have dysmorphic features that include globular eyes, micrognathia, and large ears. Affected male infants commonly have penile enlargement, whereas affected female infants often have clitoromegaly and hirsutism. In spite of hyperinsulinemia associated with glucose intolerance or diabetes mellitus, the major glucose metabolism problem for these patients is fasting hypoglycemia. Many patients with this condition do not survive past the first year of life. Unlike patients with Rabson-Mendenhall syndrome, patients with leprechaunism do not present with diabetic ketoacidosis.
Patients with Rabson-Mendenhall syndrome present during childhood with severe insulin resistance. Although patients with this disorder may present initially with fasting hypoglycemia, eventually they develop severe diabetic ketoacidosis that is refractory to insulin therapy. Patients with this condition also have acanthosis nigricans, accelerated linear growth, dystrophic nails, premature and dysplastic dentition, coarse facial features, and pineal hyperplasia.
Patients with type A insulin resistance syndrome have acanthosis nigricans and severe inherited insulin resistance in the absence of INSR autoantibodies. Patients with this syndrome tend to be lean and develop glucose intolerance. Females with this syndrome also exhibit signs of ovarian hyperandrogenism, including hirsutism, severe acne, clitoromegaly, oligomenorrhea, and infertility.
Patients with type B insulin resistance syndrome are distinguished from patients with type A insulin resistance syndrome by the presence of anti-INSR antibodies in the plasma that block insulin binding. Patients with type B insulin resistance syndrome present during adulthood with acanthosis nigricans, ovarian hyperandrogenism, and severe insulin resistance in association with signs of autoimmune disease—including alopecia areata, vitiligo, primary biliary cirrhosis, arthritis, and nephritis. Surprisingly, these patients may present with fasting hypoglycemia that may or may not be accompanied by postprandial hyperglycemia. Hodgkin disease and ataxia-telangiectasia are also associated with this syndrome. The term HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) has also been used to describe women with features of types A and B insulin resistance syndromes in association with obesity. However, this term is imprecise because many women who have been labeled as having HAIR-AN may actually have type A or B insulin resistance syndrome or severe polycystic ovary syndrome.
Mutations in the INSR have been found in all patients with leprechaunism and Rabson-Mendenhall syndrome, and in 10% to 15% of patients with type A insulin resistance syndrome. These mutations are divided into five classes. Class I mutations are frameshift or nonsense mutations that prematurely terminate translation and thus interfere with INSR synthesis. Class II mutations interfere with posttranslational processing and intracellular trafficking of the INSR. Class III mutations decrease insulin binding to the INSR. Class IV mutations are point mutations usually located on the intracellular region of the β subunit that decrease INSR TK activity. Class V mutations increase INSR degradation by increasing insulin-induced endocytosis and degradation of the receptors.
Patients with Rabson-Mendenhall syndrome and leprechaunism are homozygous or compound heterozygous for these mutations. Some patients with type A syndrome have been found to be heterozygous for dominant negative β-subunit mutations that reduce TK activity by 75%. Other patients with type A syndrome have been found to be homozygous or compound heterozygous for α-subunit mutations that interfere with receptor trafficking to the plasma membrane, β-subunit mutations that interfere with TK activity, or mutations that interfere with proreceptor cleavage into α and β subunits. Still other patients have been found to have decreased INSR mRNA levels that may be caused by a loss-of-function mutation in the INSR gene promoter. Interestingly, one patient with leprechaunism with parents with type A syndrome has been described. The proband was found to be homozygous for an INSR mutation that decreases TK activity, and the parents were found to be heterozygous for the mutation.
The insulin-like growth factor-1 receptor
The growth-promoting effects of IGF-1 are mediated by IGF1Rs. IGF1R αβ subunits are encoded by a single gene. Like the insulin receptor, an αβ half-receptor precursor is produced that then joins with a half-receptor precursor that may be encoded from the other allele to form a complete heterotetrameric IGF1R. The IGF1R has 100-fold less affinity for insulin than for IGF-1.
Patients who are heterozygous for a ring chromosome 15, which results in deletion of the IGF1R gene, have intrauterine growth retardation (IUGR) and postnatal growth failure. Other associated features include delayed bone age, mental retardation, cardiac abnormalities, cryptorchidism, and dysmorphic features that include microcephaly, triangular face, frontal bossing, hypertelorism, and brachydactyly. Similarly, IUGR and postnatal growth failure are commonly found in patients heterozygous for deletion of distal 15q that results in deletion of the IGF1R gene. Patients with deletion of distal 15q often have microcephaly, triangular facies, hypertelorism, high-arched palate, micrognathia, cystic kidneys, and lung hypoplasia or dysplasia. However, the ring chromosome and deleted area of distal 15q may lead to loss of other genes—and it is unknown to what extent absence of the IGF1R gene contributes to the complex phenotype of these patients.
Complete loss of IGF1R function is lethal in the mouse model. Nevertheless, genetic screening of 42 short children, with a history of IUGR (approximately 10% of infants with IUGR remain small), revealed a girl who was compound heterozygous for missense mutations in IGF1R (p.R108Q and p.K115N). Fibroblasts cultured from the patient had decreased IGF-1 receptor function compared with that in control fibroblasts. Moreover, in a second cohort of 50 children with short stature who had elevated circulating IGF-1 concentrations, Abuzzahab et al. identified one boy with a heterozygous nonsense mutation (R59X) in IGF1R that reduced the number of IGF-1 receptors on fibroblasts. Children with IGF1R mutations have severe IUGR, microcephaly, and short stature, with biallelic mutations having a more severe phenotype.
In addition to defective INSR function, some patients with leprechaunism and Rabson-Mendenhall syndrome are resistant to the glucose lowering or growth promotion of IGF-1 and have abnormal IGF1R function—resulting in decreased ligand binding or altered intracellular signaling. No deleterious IGF1R gene mutation has been identified in patients with these syndromes, and many patients with leprechaunism and Rabson-Mendenhall syndrome have normally functioning IGF1Rs and no evidence of IGF-1 resistance.
Until recently, no activating mutations of the IGF1R had been found. Microduplication of 15q26.3, which includes the IGF1R, is believed to be the cause of an overgrowth phenotype. Recently, a heterozygous activating mutation of the IGF1R was discovered in a patient with extreme tall stature at + 3.06 SD, 6’8”. The proband had a birth length at + 1.2 SD. His growth spurt started at age 16 years and he continued growing until age 20 years. He had very low levels of IGF-1(-3 SD), with a delayed and prolonged growth spurt associated with decreased testosterone and SHBG levels in the setting of normal LH and FSH levels. His mother and father were at + 1.1 SD and -0.3 SD in height. The variant was studied in mouse fibroblasts that lack the IGF1R and showed hyperstimulation of genes known to be regulated by the IGF1R compared with wild-type IGF1R. It also led to 50- to 70-fold increases in androgen receptor expression. The patient developed a meningioma of the sphenoid that was resected at age 40 years.
The fibroblast growth factor receptor family
There are four members of the FGFR TK family. These are FGFR1, FGFR2, FGFR3, and FGFR4. These receptors consist of a single polypeptide chain that contains an amino-terminal extracellular region, a transmembrane region, and a cytosolic region (see Fig. 3.6 ). The extracellular region contains three immunoglobulin-like domains: IgI, IgII, and IgIII (see Fig. 3.6 ). The cytosolic region contains a TK domain split into two segments (TK1 and TK2) by an intervening amino acid segment.
FGFs are a family of growth factors involved in angiogenesis, wound healing, and embryonic development. The FGFs are heparin-binding proteins and interactions with cell-surface–associated heparan sulfate proteoglycans have been shown to be essential for FGF signal transduction. FGFs are key players in the processes of proliferation and differentiation of wide variety of cells and tissues. In humans, 22 members of the FGF family have been identified, all of which are structurally related signaling molecules. As monomers, FGFs can only bind a single FGFR—forming an inactive 1:1 complex. FGFR activation by dimerization occurs when two or more FGF molecules in 1:1 complexes are linked by heparan sulfate proteoglycans.
Activation of FGFRs increases receptor TK activity. Increased TK activity leads to autophosphorylation of a tyrosine residue in the carboxy-terminal region, resulting in a binding site for the SH2 domain of phospholipase Cγ (PLCγ). Once PLCγ is bound to this site, it is phosphorylated and activated. In chondrocytes, activation of FGFR3 also induces activation of STAT1.
Fibroblast Growth Factor Receptor 1
Inactivating mutations of the FGFR1 gene are a cause of autosomal-dominant KS. Individuals with KS have anosmia and isolated hypogonadotropic hypogonadism. The FGFR1, which is located on 8p12, plays a role in olfactory and GnRH neuronal migration from the nasal placode to the olfactory bulb, and in the subsequent migration of the GnRH neurons to the hypothalamus. Before identification of these FGFR1 gene mutations, X-linked KS was thought to be caused only by inactivating mutations of the KAL1 gene. KAL1 encodes anosmin-1, the ligand for the FGFR1 receptor. Like FGFR1s, anosmin-1 plays a role in olfactory and GnRH neuronal migration to the nasal placode, and in the subsequent migration of GnRH neurons to the hypothalamus.
There is a high penetrance for anosmia and signs of hypogonadotropic hypogonadism (including lack of puberty, microphallus, and cryptorchidism) in the 10% of KS patients with X-linked KS because of KAL1 gene mutations. Female carriers of KAL1 gene mutations do not have anosmia or isolated hypogonadotropic hypogonadism. In contrast to patients with KS caused by KAL1 gene mutations, the approximately 10% of KS patients with FGFR1 gene mutations (even within the same kindred) exhibit variable phenotypes, ranging from anosmia and complete hypogonadotropic hypogonadism (characterized by cryptorchidism and microphallus in males and absent pubertal development in both genders) to anosmia or delayed puberty.
It has also been noted that in most kindreds with FGFR1 gene mutations, females present with more mild KS phenotypes than males. Female carriers may even be asymptomatic. Because the KAL1 gene is located on the X chromosome, females may produce more anosmin-1 than males. Thus a possible explanation for milder KS phenotypes in females with FGFR1 gene mutations may be that the increased anosmin-1 levels in females may lead to increased anosmin-1–induced activation of the mutant FGFR1s that may partially compensate for the mutation. Interestingly, missing teeth and cleft palate are not an uncommon finding in individuals with KS resulting from FGFR1 gene mutations, whereas unilateral renal agenesis and bilateral synkinesia are associated with KS arising from KAL1 gene mutations.
Fibroblast Growth Factor Receptors 2–4
A diverse group of skeletal disorders are caused by activating mutations in the FGFR1 gene, as well as in the related genes encoding FGFR2 and FGFR3. In general, gain-of-function mutations in FGFR1 and FGFR2 cause most of the syndromes involving craniosynostosis, whereas the dwarfing syndromes are largely associated with FGFR3 mutations. Osteoglophonic dysplasia is a “crossover” disorder that has skeletal phenotypes usually associated with FGFR1, FGFR2, and FGFR3 mutations. Osteoglophonic dysplasia is caused by missense mutations in highly conserved residues comprising the ligand-binding and transmembrane membranes of FGFR1, thus defining novel roles for this receptor as a negative regulator of long bone growth. Gain-of-function mutations in genes encoding the other FGF receptors cause related skeletal disorders: Pfeiffer syndrome (activating mutations of FGFR1 and FGFR2), Crouzon syndrome (gain-of-function FGFR2 mutations), Crouzon syndrome with acanthosis nigricans (an FGFR3 mutation), Apert syndrome (FGFR2 mutations), and craniosynostosis (gain-of-function FGFR3 mutations). Several autosomal-dominant short-limb dwarfism syndromes—including achondroplasia, severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN), hypochondroplasia, and three types of platyspondylic lethal skeletal dysplasias (PLSD) (thanatophoric dysplasia I [TDI], thanatophoric dysplasia II [TDII], and San Diego types [PLSD-SD])—are often caused by heterozygous constitutively activating FGFR3 gene mutations.
Individuals with achondroplasia have activating mutations in the transmembrane domain of FGFR3, with the p.Gly380Asn found in more than 95% of achondroplastic patients. From 40% to 70% of individuals with hypochondroplasia have an activating p.Asn540Lys mutation in the TK1 domain. All individuals with TDII have an activating p.Lys650Glu mutation in the activating loop of the TK2 domain, and more than 90% of individuals with TDI and PLSD-SD have FGFR3 mutations. Patients with SADDAN have an activating mutation in the same codon as patients with TDII. Instead of the p.Lys650Glu mutation associated with TDII, patients with SADDAN have a Lys650Met mutation. However, unlike patients with TDII patients with SADDAN do not have craniosynostosis and a cloverleaf skull—and often survive past childhood.
The FGFR3 gene is primarily expressed in endochondral growth plates of long bones, brain, and skin pre- and postnatally. Constitutional activation of FGFR3s in chondrocytes leads to growth arrest and apoptosis. In addition, constitutive activation of FGFR3s is also postulated to alter neuronal migration because patients with SADDAN, TDI, and TDII have neurologic abnormalities that may include developmental delay, paucity of white matter, polymicrogyria, dysplastic temporal cortex, dysplasia of nuclei, and neuronal heterotopia. Furthermore, constitutive activation of FGFRs in skin fibroblasts and keratinocytes is thought to cause the acanthosis nigricans seen in patients with SADDAN and Crouzon syndrome with acanthosis nigricans. However, it is not yet known why some activating FGFR3 mutations affect the skeletal system, central nervous system, and the skin, whereas other activating FGFR3 mutations only affect the skeletal system.
Loss-of-function mutations in FGFR3 also have been associated with human disease. An uncommon syndrome characterized by camptodactyly, tall stature, scoliosis, and hearing loss (CATSHL syndrome) has been associated with a heterozygous missense mutation (p.R621H) in the TK domain and partial loss of FGFR3 function. These findings indicate that abnormal FGFR3 signaling can cause human anomalies by promoting, as well as inhibiting, endochondral bone growth.
Nuclear receptors
Using a phylogenetic tree based on the evolution of two highly conserved nuclear receptor domains (the DNA-binding C domain and the ligand-binding E domain), Laudet divided nuclear receptors into six related subfamilies. Subfamily 0 contains receptors, such as the embryonic gonad (EGON) and DAX1 receptors, that do not have a conserved C or the E domain ( Fig. 3.8 ). Subfamily 1 includes the peroxisome proliferator-activated retinoic acid, thyroid hormone, and vitamin D 3 receptors. Subfamily 2 includes the hepatocyte nuclear factor-4α (HNF4A) and retinoid X receptors (RXRs). Subfamily 3 contains the steroid receptors. Evidence suggests that subfamily 3 (which includes the glucocorticoid, androgen, progesterone, and mineralocorticoid receptors) rapidly evolved from a common steroid receptor gene about 500 million years ago.
Subfamilies 4 to 6 contain several nuclear receptors, namely NR4A1-3, NR5A1-2, and NR6A1. The nomenclature is as follows: nuclear receptor subfamily 4, group A, and members 1 for NR4A1. NR5A1 is also known as steroidogenic factor-1 or SF-1 .
General Structure of the Nuclear Receptors
Nuclear receptors are made up of four domains: A/B, C, D, and E ( Fig. 3.9 ). Supporting the notion that nuclear receptor subfamilies are derived from a common ancestral orphan receptor, the C and E domains are highly conserved among the subfamilies. Mutations of several nuclear receptors are associated with endocrine disorders ( Table 3.5 ).
| Receptor | Germ-Line Mutation | Endocrine Disorder |
|---|---|---|
| Thyroid Hormone Receptor β (TRβ) | Inactivating mutations (heterozygous and homozygous) | Generalized resistance to thyroid hormones |
| Vitamin D 3 receptor | Inactivating mutations (homozygous) | Vitamin D 3 resistance |
| PPARγ2 | Inactivating mutations (heterozygous) | Obesity or early-onset type 2 diabetes mellitus |
| HNF-4 | Inactivating mutations (heterozygous) | Maturity-onset diabetes of the young (MODY) type 1 Hyperinsulinemic hypoglycemia in the newborn period followed in later life with MODY1 in some patients |
| Glucocorticoid receptor | Inactivating mutations (heterozygous) | Glucocorticoid resistance |
| Androgen receptor | Inactivating mutations (X-linked recessive) | Androgen insensitivity syndrome, Kennedy disease |
| Estrogen receptor α (ER α) | Inactivating mutations (homozygous) | Tall stature and incomplete epiphyseal fusion |
| Mineralocorticoid receptor | Inactivating mutations (heterozygous, homozygous) | Pseudohypoaldosteronism type 1 |
| Activating mutations (homozygous) | Syndrome of apparent mineralocorticoid excess | |
| DAX1 | Inactivating mutations (X-linked recessive) | X-linked adrenal hypoplasia congenita |
The A/B domain is located at the amino-terminal and contains the activation function 1 (AF-1)/τ 1 domain. The AF-1/τ 1 domain regulates gene transcription by interacting with proteins (such as the Ada and TFIID complexes) that induce transcription. This transactivation function of the AF-1/τ 1 domain is not dependent on binding of the nuclear hormone receptor to its ligand and is not specific in its choice of DNA target sequences. Thus specificity of action of the nuclear hormone receptor is determined by the function of other nuclear hormone receptor domains.
The C domain has characteristics that help to confer specificity of action on each nuclear hormone receptor. This domain consists of two zinc-finger motifs responsible for the DNA-binding activity of the receptor and the selection of dimerization partners. Each zinc-finger module consists of a zinc ion surrounded by the sulfurs of four cysteine residues, resulting in a tertiary structure containing helices. The P-box lies near the cysteines of the first zinc finger and contains the three to four amino acids responsible for specificity of binding to response elements. The D-box consists of a loop of five amino acids attached to the first two cysteines of the second zinc finger that provides the interface for nuclear receptor dimerization.
The D “hinge” domain contains nuclear localization signals and contributes to the function of the adjacent C and E domains. Thus the amino-terminal portion of the domain contributes to DNA binding and heterodimerization and the carboxy-terminal portion contributes to ligand binding. The nuclear localization signal plays a particularly important role in the function of glucocorticoids and mineralocorticoid receptors because these receptors bind their ligand in the cytoplasm and must then localize to the nucleus to alter gene transcription.
The E domain is known as the ligand-binding domain ( LBD ) or the hormone-binding domain . In addition to ligand binding, the E domain has effects on dimerization and transactivation. The LBD consists of 11 to 12 α helices (named H1 through H12) and contains a ligand-binding pocket that is made up of portions of some of the different helices. For example, the thyroid receptor (TR) LBD has a ligand-binding cavity that includes components from H2, H7, H8, H11, and H12. The contribution of different parts of LBD to the ligand-binding pocket accounts for the finding that mutation of single–amino-acid molecules in different helices of the LBD can interfere with ligand binding.
Unlike the AF-1/τ 1 transcriptional activating factor, the E domain activation factor 2 (AF2-AD) requires ligand binding to function (see Fig. 3.9 ). Often, when the receptor is not bound by its ligand, corepressor complexes simultaneously bind the LBD and transcriptional machinery consisting of protein complexes that place transcription factors on nucleosome binding sites (see Fig. 3.9 ). The corepressor complexes then suppress gene transcription by using histone deacetylases to compact the nucleosomes into inaccessible structures (see Fig. 3.9 ). Ligand binding induces structural rearrangements in the E domain that lead to release of these corepressor complexes from the transcriptional machinery and the LBD, and exposure of the transcriptional machinery and the LXXLL motif of the AF2-AD to coactivator complexes (see Fig. 3.9 ). These coactivator complexes have histone acetyltransferase activity that acts to relax nucleosome structures, enabling transcription factors to access nucleosome binding sites (see Fig. 3.9 ).
Most nuclear receptors are capable of binding their hormone response element and repress transcription when they are not bound by their ligand. However, in the absence of ligand, steroid receptors are bound to a complex of heat-shock proteins instead of their response element and do not appear to repress transcription.
Agonists and antagonists have different effects on the interaction between the ligand binding pocket and AF2-AD. For example, when 17β-estradiol binds to the estrogen receptor, the position of the AF2-AD containing H12 is altered so that coactivators can access the LBD-binding coactivator binding site. However, when the estrogen antagonist raloxifene binds at the same site, the coactivator binding site on H12 remains blocked by other portions of H12.
Although some nuclear receptors are fully active when bound as monomers to DNA, the hormone receptors in the nuclear receptor superfamily are most active when bound as homodimers or heterodimers (see Fig. 3.9 ). RXRs, HNF 4, and the steroid hormone receptors can bind DNA as homodimers or heterodimers. The α isoform of the estrogen receptor (ESR1) is particularly promiscuous, and is able to heterodimerize with HNF4A and retinoic acid receptors, the β isoform of the estrogen receptor (ESR2), RXR, and the thyroid hormone receptors. As a homodimer, RXR binds the dINSRect repeat 1 (DR1). It can also join the thyroid, vitamin D 3 , and peroxisome proliferator–activated receptors to form heterodimers.
Interestingly, it has also been suggested that some steroid hormones also act on transmembrane receptors—and these interactions may be responsible for the acute cellular effects of steroids. Progesterone has been shown to interact with the G protein–coupled uterine oxytocin, nicotinic acetylcholine, γ-aminobutyric acid A , N -methyl- D aspartate, and sperm cell membrane progesterone receptors. Cell membrane estrogen and glucocorticoids receptors have also been identified.
Subfamily 1 nuclear receptors: thyroid hormone, vitamin d 3 , and peroxisome proliferator–activated receptors
Thyroid Hormone Receptors
The two thyroid hormone receptor (THR) isoforms—thyroid hormone receptor α (THRA) and thyroid hormone receptor β (THRB)—are encoded by different c-erbA genes on chromosomes 17 and 3, respectively. Alternate splicing leads to expression of TRα1, TRα2, TRβ1, and TRβ2 with different tissue distributions. TRα2 does not bind thyroid hormone and its function is not understood. TRα1 is the predominant subtype in cardiac and skeletal muscle, bone, gastrointestinal tract, and the central nervous system. TRβ1 is the predominant subtype in kidney and liver, whereas TRβ2 is expressed in hypothalamus and pituitary, as well as retina and cochlea. THRs that are not occupied by the thyroid hormone triiodothyronine (T 3 ) exist as homodimers or heterodimers, with RXRs that are attached to DNA thyroid hormone response elements in association with corepressor proteins. Thyroid hormone binding induces the release of the corepressors from the THR. A coactivator, steroid receptor coactivator-1 (SRC-1), is then able to attach to the THR—enabling activation of transcription.
Generalized resistance to thyroid hormones (GRTH) can be autosomal recessive or autosomal dominant, as the presence of a single normal THRB allele is sufficient for normal receptor function. Autosomal dominant GRTH is caused by the presence of an abnormal THRB that interferes with the function of the normal receptor in a dominant-negative fashion. The prevalence of dominant GRTH, based on newborn screening, was reported to be 1:40,000 live births. Patients with this syndrome have an impaired receptor response to T 3 . They have elevated T 3 , reverse T 3 , and thyroxine (T 4 ) levels with slightly high TSH levels. Consistent with the TSH elevation and goiter, serum thyroglobulin levels tend to be elevated and radioiodine uptake high. Unlike autoimmune thyrotoxicosis, the T 3 : T 4 ratio is normal. The clinical manifestations are variable including some hyperthyroid and some hypothyroid symptoms. Findings include goiter in 65% to 85% of those affected, hyperactivity in 33% to 68%, tachycardia in 33% to 75%, failure to thrive, raised metabolic rate, low bone density, hearing defects, learning disabilities, developmental delay, and delayed bone age. GRTH can be detected by newborn screening if the program measures T4, in addition to or in lieu of TSH only, as T 4 would be elevated and TSH would be elevated. The combination of findings normally associated with hyperthyroidism and hypothyroidism is caused by differential expression of TRα versus TRβ. The hypothalamus and pituitary express TRβ and this leads to elevated TSH levels. Cardiac tissues express TRα and this leads to tachycardia as elevated T 4 and T 3 act on the normal TRα receptor.
Mutations have been found in the D and E domains of THRBs of GRTH patients. These mutations alter ligand binding or transactivation. However, most mutant THRBs retain the ability to repress transactivation of target genes through interactions with corepressors. Some of the GRTH mutant receptors continue to associate with corepressors and are unable to bind the coactivator SRC-1 even when bound by T 3 . Thus mutant THRBs have a dominant negative effect in the heterozygous state because they are able to interfere with the function of wild-type receptors by repressing transcription of target DNA.
Patients who carry biallelic loss-of-function mutations of THRB demonstrate more severe clinical abnormalities than patients with heterozygous dominant negative mutations. One patient with a deletion of both THRB alleles presented with deaf mutism, dysmorphic features, and stippled epiphyses. Another patient homozygous for a THRB mutant (“kindred S receptor”), with an amino acid deletion in the ligand-binding domain, presented with mental retardation, very delayed bone age, and very elevated T 3 and T 4 levels. Heterozygous carriers of the kindred S receptor mutation have milder clinical manifestations of GRTH because this mutant THRB retains corepressor activity and thus has dominant negative effects.
Mutations in THRA were not identified until recently. The first described proband is heterozygous for a nonsense mutation (p.E403X) that inhibits the wild-type receptor in a dominant negative fashion. The patient had hypothyroid features (growth retardation, developmental delay, skeletal dysplasia, and constipation) that reflect the distribution of target tissues in which THRA is expressed. This is associated with borderline low or normal T 4 and free T 4 , low reverse T 3 , borderline high or normal T 3 and free T 3 , a low T 4 : T 3 ratio, and normal TSH. After thyroxine treatment, normalizing T 4 levels, T 3 levels became elevated. It has been suggested that this may be related to increased type 1 deiodinase activity (which converts T 4 to T 3 ) and reduced type 3 deiodinase activity (which converts T 4 to reverse T 3 and T 3 to T 2 ). The mutant receptor failed to bind radiolabeled T 3 and failed to activate a thyroid hormone responsive reporter gene and inhibited the activity of the wild-type receptor. The second report described a one base insertion in a father and a daughter, causing a frameshift and premature termination at codon 406. Analyses of the mutant receptor in vitro, after expression in cultured cells, revealed that the mutant receptor fails to respond to stimulation by T 3 and exerts a strong dominant negative effect on the wild-type receptor. This and other subsequent reports identified more clinical features, such as macrocephaly, dysmorphic facies (round, flat face with course features) increased birth length and weight, anemia, and slightly elevated cholesterol. Several reports have recently documented different mutations, including missense mutations with partially active receptors. These studies demonstrated that the severity of clinical findings correlated with the degree of receptor impairment. The p.A263V mutation exhibited impaired transcription at low T 3 concentrations, but was active at higher T 3 concentrations and was associated with a milder phenotype. Another mutation (p.N359Y), which affects both TRα1and TRα2, was associated with additional clinical features, namely, micrognathia, clavicular agenesis, metacarpal fusion, syndactyly, hyperparathyroidism, and chronic diarrhea.
Vitamin D Receptor
Severe rickets, hypocalcemia, secondary hyperparathyroidism, and increased 1,25-dihydroxyvitamin D (calcitriol) levels occur in patients with the autosomal recessive syndrome of “vitamin D resistance.” These patients have defective vitamin D receptors (VDRs). Mutations causing this syndrome have been found in the zinc fingers of the DNA-binding domain (C domain), leading to decreased or abolished receptor binding to regulatory elements of target genes. Causative mutations have also been found that lead to the production of receptors that have decreased or abolished ability to bind calcitriol and heterodimerize with RXRs, which are required for the VDR to maximally transactivate target genes.
Less severe mutations in the VDR are associated with decreased gastrointestinal calcium absorption and bone mineral density, even during childhood, and an increased risk for osteoporosis and fractures. However, it has not been possible to replicate these findings in some ethnic groups. Thus other factors (such as estrogen receptor genotype, dietary calcium, and age) probably contribute to the effects of VDR polymorphisms on bone mineral metabolism. Certain VDR polymorphisms are associated with decreased pre- and postnatal linear growth.
Other important associations have been found with VDR polymorphisms. Homozygous polymorphisms have been reported to be associated with primary hyperparathyroidism. In addition, the presence of certain VDR alleles is associated with increased risk for the development of early-onset periodontal disease. Conversely, absence of such alleles has been associated with familial calcium nephrolithiasis. Absence of certain alleles may be a risk factor for the development of sarcoidosis. However, the presence of these alleles is associated with hypercalciuria and nephrolithiasis and increased risk in women for the development of metastatic breast cancer. VDR polymorphisms have also been associated with increased susceptibility to psoriasis, tuberculosis, leprosy, and other infections.
Peroxisome Proliferator-Activating Receptor γ
Peroxisome proliferator-activating receptor γ (PPARγ) has a role in regulating adipocyte differentiation and metabolism. This formerly orphan receptor was adopted by the ligand prostaglandin J2. Mutations in the gene encoding PPARγ cause familial partial lipodystrophy type 3 ( FPLD3 ).
Patients with FPLD3 exhibit fat loss in the arms and legs, severe insulin resistance, early-onset type 2 diabetes mellitus, and severe hypertriglyceridemia. Some heterozygous mutations have a dominant negative effect on the wild-type receptor (V290M and P467L). These mutations lead to amino acid substitutions that disturb the orientation of H12 in the E domain, leading to decreased ligand-dependent transactivation by AF-2/AD and coactivator recruitment. Other mutations are point mutations that cause FPLD3 simply because of haploinsufficiency without a dominant negative effect. In addition, a frameshift mutation that causes a similar phenotype was identified. Another mutation in PPARγ expanded the phenotype to include muscular, immune, and hematologic features. This mutation is thought to induce a conformational change affecting transcriptional activation by the receptor.
A missense mutation leading to a p.Pro115Gln substitution near a site of serine phosphorylation at position 114 that suppresses transcriptional activation in PPARγ2 was found in some morbidly obese patients. The p.Pro115Gln substitution interferes with phosphorylation of the serine at position 114, leading to increased transcriptional activation by PPARγ2—which in turn leads to increased adipocyte differentiation and triglyceride accumulation.
Subfamily 2 nuclear receptors: hepatocyte nuclear factor and retinoid x receptors
This subfamily includes the HNF receptors and the RXRs. RXRs form heterodimers with other nuclear receptors (including the estrogen, vitamin D, and thyroid hormone receptors) and with PPARγ (RXRs are discussed elsewhere in this chapter).
Hepatocyte Nuclear Factor Receptors
Alteration of another orphan nuclear receptor, HNF4A, also causes an endocrine disorder. Mutations of the HNF4A gene on chromosome 20 that alter the ligand-binding domain (E domain) or the DNA-binding domain (C domain) have been found in patients with a form of monogeneic diabetes, also known as maturity-onset diabetes of the young type 1 (MODY1). Patients with MODY usually develop diabetes mellitus by the end of the third decade of life. They have a defect in glucose-mediated stimulation of insulin secretion. Several studies have now described a biphasic phenotype of neonatal hyperinsulinism and macrosomia, with later onset of diabetes in patients with HNF4A mutations. In most cases, the hyperinsulinism is transient but can be persistent, requiring diazoxide treatment.
Patients who are heterozygous for the p.R76W HNF4A mutation also develop a Fanconi-Bickel like syndrome in addition to neonatal hyperinsulinism and macrosomia. The patients manifest the typical proximal tubulopathy of Fanconi-Bickel syndrome, including generalized aminoaciduria, low-molecular-weight proteinuria, glycosuria, hyperphosphaturia, and hypouricemia, plus additional features, including nephrocalcinosis, renal impairment, hypercalciuria with relative hypocalcemia, and hypermagnesaemia. The Fanconi-Bickel like phenotype has been recapitulated in a mouse model in which genetic ablation of Hnf4a was targeted to the kidney, indicating that Hnf4a is required for the formation of differentiated proximal tubules and that loss of Hnf4a decreased the expression of proximal tubule-specific genes. The p.R76W mutation occurs in the DNA-binding domain of HNF4A, and has been hypothesized to cause defective interaction with major regulatory genes; it remains unknown why patients with other HNF4A mutations do not develop the same renal phenotype.
Carriers of a glycine-to-serine substitution in codon 115 in the DNA-binding domain (C domain) appear to be at increased risk for developing low-insulin diabetes mellitus. Hepatocyte nuclear factors 3 (HNF-3α, -3β, and -3γ) are also regulators of the early-onset type 2 diabetes genes HNF1A , HNF4A , and IPF-1/PDX-1 —which are associated with MODY types 3, 1, and 4, respectively.
Subfamily 3 nuclear receptors: the steroid receptors and glucocorticoid, androgen, estrogen, and mineralocorticoid receptors
Glucocorticoid Receptors
Glucocorticoids strongly influence cardiovascular tone; have actions on liver, muscle, and adipose tissue; and exert potent antiinflammatory and immunosuppressive effects. Glucocorticoids are important in growth and development, as well as behavior and cognition. They influence multiple cellular processes, including proliferation, differentiation, and apoptosis. All of these actions are mediated by the glucocorticoid receptor (GR), which is a ligand activated transcription factor influencing transcription of approximately 20% of the genome. The gene for the glucocorticoid receptor is known as NR3C1 . NR3C1 is on chromosome 5 and consists of 10 exons. Multiple isoforms of the GR exist because of alternative splicing, insertions, deletions, and alternative translation initiation. The two main forms are hGRα and hGRβ. hGRβ does not bind natural or synthetic glucocorticoids; is expressed in few cell types, such as neutrophils and epithelial cells; and may exert inhibitory effects on the main glucocorticoid receptor hGRα. hGRα is ubiquitously expressed in all tissue but the suprachiasmatic nuclei of the hypothalamus.
Mutations in glucocorticoid receptors cause primary generalized glucocorticoid resistance (PGGR) or primary generalized glucocorticoid hypersensitivity (PGGH). PGGR is clinically characterized by the presence of elevated plasma cortisol and ACTH levels, accompanied by the effects of hyperaldosteronism and hyperandrogenism in the absence of striae or central fat deposition. ACTH excess leads to adrenocortical hyperplasia, increased cortisol secretion, and increased production of adrenal steroids with androgenic (androstenedione, dehydroepiandrosterone [DHEA], DHEA-S) and mineralocorticoid activity (cortisol, deoxycorticosterone and corticosterone). The clinical spectrum is broad, ranging from asymptomatic to severe hyperandrogenism (characterized by severe acne, hirsutism, irregular menses, and infertility), mineralocorticoid excess (characterized by hypokalemic alkalosis and hypertension), and fatigue. Girls may present with ambiguous genitalia and both girls and boys may present with isosexual precocity. Clinical manifestations of glucocorticoid deficiency are infrequent and largely limited to fatigue. However, there were reports of childhood hypoglycemia.
Both homozygous and heterozygous mutations have been described. Heterozygous mutations that cause PGGR generally do so by exerting a dominant negative effect on the wild-type receptor.
ACTH-secreting pituitary macroadenomas can also be caused by a frameshift mutation in the GR gene that interferes with signal transduction. Patients with this mutation manifest the symptoms of glucocorticoid resistance. The tumor develops as a result of impaired negative feedback regulation by glucocorticoids on the hypothalamic-pituitary axis.
PGGH was described in a 43-year-old female who presented with visceral obesity, hypertension, hyperlipidemia, and type 2 diabetes, and was found to have a mutation in the GR (D401H) that demonstrated increased transactivation of glucocorticoid responsive genes. The patient had evidence of glucocorticoid resistance in the hypothalamus–pituitary gland–adrenal gland axis and hypersensitivity at the vasculature, adipose tissue, and liver. This was associated with an elevated ACTH and am cortisol but normal urine free cortisol.
Androgen Receptors
The human androgen receptor ( AR ) gene is located on the X chromosome. Known disorders characterized by AR dysfunction caused by AR gene mutations are only expressed in patients with a 46 XY karyotype. These mutations may be transmitted from an asymptomatic carrier mother or can be de novo.
More than 1000 mutations have been described in the AR gene, with more than 500 mutations causing androgen insensitivity syndrome (AIS). The phenotype of this syndrome can vary in severity and has been divided into mild, partial, and complete forms (MAIS, PAIS, and CAIS). CAIS is characterized by intraabdominal testes, absence of mullerian structures, absence of androgen-induced body hair, such as pubic and axillary hair, and a female appearance. PAIS refers to individuals with ambiguous external genitalia with an enlarged clitoris or microphallus and patients with Reifenstein syndrome. Reifenstein syndrome is characterized by severe hypospadias with scrotal development and severe gynecomastia. MAIS refers to patients with AR mutations who are otherwise normal phenotypic males who present with adolescent gynecomastia or later infertility.
The phenotypic heterogeneity of AIS is caused by the varied locations of the mutations causing AIS. Functional consequences of each mutation causing AIS relate to the function of the domain in which the mutation is located. However, the degree of impairment of mutated receptor function in in-vitro studies does not always correlate with the phenotypic severity of the syndrome.
Mutations in exons that code for the AR hormone-binding domain decrease hormone-binding affinity. However, these mutations do not abolish the hormone-binding capability of the receptor. Thus patients with these mutations usually present with PAIS or occasionally with CAIS. Patients with mutations in the hormone-binding domain do not appear to respond to treatment with high doses of testosterone. Mutations in the DNA-binding domain lead to failure of target gene regulation. Thus patients with these mutations usually manifest the CAIS.
CAIS is also caused by a point mutation that results in a premature termination codon (p.Gln340ter), with apparent initiation of translation that is downstream of the termination codon. In vitro studies showed that the truncated AR is expressed at reduced levels. Numerous other mutations have been described that cause truncation or deletion of the AR and complete AIS. Two patients with ambiguous genitalia and partial virilization were found to be mosaic for mutant ARs.
Some patients with Reifenstein syndrome have been found to have a mutation in the DNA-binding domain that abolishes receptor dimerization. Other patients have been found to have a mutation in a different area of the DNA-binding domain that does not affect receptor dimerization, or to have mutations in the hormone-binding domain in the E domain. The Ala596Thr mutation in the D-box area of the DNA-binding domain has been associated with an increased risk of breast cancer.
AIS is also a feature of Kennedy disease, which is an X-linked recessive condition causing spinal and muscular atrophy. This condition is caused by extension of a poly-CAG segment in the AR gene exon that codes for the N-terminus of the AR, leading to an increased number of glutamine residues in the A/B domain. ARs with a polyQ region increased to 48 glutamine residues accumulate abnormally in transfected cells because of misfolding and aberrant proteolytic processing. Because polyQ extension does not completely abolish transactivation, patients with Kennedy disease exhibit a mild partial AIS phenotype consisting of normal virilization accompanied by testicular atrophy, gynecomastia, and infertility.
Gain-of-function mutations in the AR have been described in prostate, breast, testicular, liver, and laryngeal cancers.
Estrogen Receptors
Two major full-length ESR isoforms have been identified in mammals. Estrogen receptor α (ESR1) was discovered first and mediates most of the known actions of estrogens. ESR1 is expressed primarily in the uterus, ovaries, testes, epididymis, adrenal cortices, and kidneys. Estrogen receptor β (ESR2) was discovered in 1996. ESR1 and ESR2 share 95% and 50% homology in the DNA-binding domain (DBD) and LBD, respectively. There is little homology in the amino-terminal between the two isoforms. ESR2 is expressed primarily in the uterus, ovaries, testes, prostate, bladder, lung, and brain. Although ESR2 has a high affinity for estrogens, it has less transactivating ability than ESR1 and has not yet been found to be involved in any pathologic condition.
There is evidence supporting the existence of other functional ESRs. Some of these putative ESRs localize to the cell membrane instead of, or in addition to, the nucleus. A 46-kDa amino-terminal truncated product of ESR1, named ER46, localizes to the cell membrane and mediates estrogen actions that are initiated at the cell membrane. Another of these putative ESRs has been named ER-X and is postulated to be a GPCR that localizes to the cell membrane. Another putative receptor is the aptly named heterodimeric putative estrogen receptor (pER), which has been found on the cell and nuclear membranes. The pER acts as a serine phosphatase. Five other estrogen-binding proteins have also been identified, and at least three of them localize to the cell membrane.
It had been thought that androgens provide the principal signals for closure of the epiphyses during puberty. In 1994, however, extensive studies of a 28-year-old man, with incomplete closure of epiphyses and tall stature, demonstrated that he was homozygous for a premature termination mutation of codon 157 in exon 2 of the ESR1 gene. The patient had continued linear growth despite otherwise normal pubertal development, demonstrating that the ESR mediates epiphyseal closure. Expression of this gene leads to the production of a nonfunctional ESR1 lacking both the DNA- and hormone-binding domains. He was also found to have increased estradiol levels, impaired glucose tolerance with hyperinsulinemia, and decreased bone density.
Further strengthening the association between ESR1 and epiphyseal closure is the observation that women with ESR1-positive breast cancer and a mutation in the B domain (B’ allele) of ESR1 have an increased incidence of spontaneous abortion and tall stature. These associations were not found in female carriers of the allele without breast cancer or with ESR1-negative breast cancer. Thus a second (as yet undiscovered) mutation is likely to play a role in the development of tall stature and spontaneous abortions in female carriers with ESR1-positive breast cancer.
Mineralocorticoid Receptors
Mineralocorticoid resistance is also known as pseudohypoaldosteronism (PHA). Both sporadic and familial cases with either autosomal-dominant or autosomal-recessive cases have been reported. Clinical presentation of patients with PHA ranges from asymptomatic salt wasting; to growth failure; to chronic failure to thrive, lethargy, and emesis; to life-threatening dehydration accompanied by severe salt wasting. Patients with the severe forms of PHA typically present within a year of birth and may even present in utero with polyhydramnios because of polyuria. Biochemically, the condition is characterized by urinary salt wasting, hyponatremia, elevated plasma potassium, aldosterone, and renin activity, and urinary aldosterone metabolism that are unresponsive to the mineralocorticoid treatment.
Two forms of PHA are recognized (types I and II). Autosomal recessive (generalized) PHAI results from mutations in the epithelial sodium channel (ENaC), whereas autosomal dominant (renal) PHAI is caused by mutations in the mineralocorticoid receptor (MR). PHAII is the result of mutations in a family of serine-threonine kinases known as WNK1 and WNK4 , which are downstream of aldosterone action. PHAI is characterized by renal tubular mineralocorticoid resistance, whereas PHAII results from mineralocorticoid resistance in the kidney, intestine, or salivary or sweat gland and is also known as Gordon syndrome . Patients with these conditions present with hyperkalemia that only responds to treatment with nonchloride ions, such as bicarbonate or sulfate, which increase delivery of sodium to the distal tubule. In general, patients with autosomal dominant PHA1 have a mild salt-wasting syndrome and improve with age, whereas patients with autosomal recessive PHA1 have severe salt wasting and hyperkalemia, as well as increased sweat and salivary sodium and frequent respiratory tract infections that fails to improve with age.
More than 50 different mutations of the MR causing autosomal dominant PHAI have been described (all heterozygous). There is clear heterogeneity of clinical manifestations within families. Haploinsufficiency caused by mRNA or protein degradation is clearly sufficient to cause autosomal dominant PHAI. Yet some mutations have been shown to exert dominant negative effects on the wild-type receptor.
Genetic disorders that are associated with increased activity of the MR cause severe early-onset hypertension. One such condition, characterized by excessive MR action, is termed the syndrome of apparent mineralocorticoid excess . Patients with this autosomal-recessive condition can exhibit pre- and postnatal growth failure, hypervolemic hypertension, medullary nephrocalcinosis, and hypokalemic metabolic alkalosis accompanied by hyporeninemic hypoaldosteronism. Patients may also be asymptomatic and exhibit only biochemical abnormalities. Patients with this syndrome also have increased serum and urinary cortisol-to-cortisone ratios. This syndrome is caused by mutations in the 11 β-hydroxysteroid dehydrogenase type 2 ( HSD11B2 ) gene that reduce enzymatic activity of the protein. The 11 β-HSD2 converts the active glucocorticoid cortisol to inactive cortisone; in contrast the type I isoform, encoded by the separate HSD11B1 gene, has both 11-β dehydrogenase activity and 11-oxoreductase activity and can also convert cortisone to cortisol. The type I isoform is ubiquitously expressed, whereas the type II isoform has more restricted expression and is highly expressed in the kidney, where it is necessary to protect the renal MR from normally higher serum concentrations of cortisol, allowing aldosterone to regulate sodium homeostasis. Thus decreased activity of 11 β-HSD2 increases cortisol levels in kidney cells expressing MR tissues, leading to increased binding and activation of MRs by cortisol.
A transient form of mineralocorticoid resistance, probably because of abnormal maturation of aldosterone receptor function, also exists. This variant of PHA is known as the syndrome of early-childhood hyperkalemia . Children with this disorder present with failure to thrive or linear growth failure accompanied by hyperkalemia and metabolic acidosis. This condition resolves spontaneously by the second half of the first decade of life.
Subfamily 0 nuclear receptors: dax1
Subfamily 0 nuclear receptors include DAX1. DAX1 plays a role in the regulation of steroid, mullerian-inhibiting substance, and gonadotropin production.
DAX1
The dosage-sensitive sex-reversal adrenal hypoplasia congenital critical region on the X chromosome gene 1 ( DAX1 ) is an orphan nuclear receptor because its ligand has not yet been identified. It has homologies in the E domain to other orphan receptors, including RXRs. However, DAX1 has an unusual DNA-binding C domain that contains a tract of amino acid repeats instead of zinc-finger motifs. DAX1 inhibits SF-1-mediated transcription. SF-1 is another orphan nuclear receptor that regulates transcription of adrenal and gonadal steroid hydroxylases, mullerian-inhibiting substance, and gonadotropin genes.
DAX1 gene mutations have been identified that cause X-linked adrenal hypoplasia congenita. Patients with this condition have congenital adrenal insufficiency and are therefore deficient in glucocorticoid, mineralocorticoid, and androgen production. Some 40% of affected male patients present in the first 2 months of life with a salt-wasting adrenal crisis, and the remainder present later in childhood with mineralocorticoid insufficiency, resembling aldosterone synthase deficiency or PHA. Those who present with adrenal crisis can have hyperkalemia, hyponatremia, hypotension, or cardiac arrest, because of both mineralocorticoid and glucocorticoid deficiency in the setting of normal 17-hydroxyprogesterone levels. Patients may have normal cortisol levels. Imaging of the adrenal by any modality at this age is not helpful, as the condition affects the definitive zone but not the fetal zone of the adrenal. The fetal zone is quite large at birth and does not atrophy until 6 to 12 months of age. Imaging cannot differentiate the fetal from the definitive zones and reports a normal adrenal gland in the first few months of life. Imaging after the first year of life would be expected to show a hypoplastic adrenal. Gonadotropin deficiency and azoospermia also occur in these patients but this does not present until puberty. Patients have findings consistent with hypogonadotropic hypogonadism (because of a combined hypothalamic/pituitary defect) and may have absent pubertal development or puberty that arrests at around Prader stage 3. Female carriers may have delayed puberty. All mutations that have been found to cause X-linked congenital adrenal hypoplasia are either located in or prevent transcription of the area of the E domain that inhibits SF-1-mediated transcription. Thus DAX1 mutations may cause X-linked congenital adrenal hypoplasia by altering SF-1 regulation of gonado- and adrenogenesis. DAX1 deletion may also occur in the setting of the contiguous gene deletion syndrome, resulting in complex glycerol kinase deficiency (cGKD) if individuals have deletions extending from the GK gene into the Duchene muscular dystrophy ( DMD ) gene, or involving a significant extension telomeric from DAX1.
Duplications of Xp that include DAX1 in genetic males with XY karyotype lead to sex reversal. DAX1 overexpression represses all aspects of male differentiation beginning with the urogenital ridge progressing to a bipotential gonad, under the influence of WT1 and SF1, to differentiation into a testis under the influence of SF1, SOX9, SRY, and AMH and leads to female external genitalia with variable female internal organs from absent ovaries, uterus, and cervix to normal ovaries with rudimentary mullerian structures. Duplications in this region of the X chromosome are also associated with developmental delay, low-set ears, cleft palate, clinodactyly, hypotonia, thin upper lip, simian creases, and cardiac abnormalities.
Summary
Understanding of receptors that transduce or influence hormone action has increased dramatically. As molecular biology techniques improve, it is expected that knowledge of receptor action will continue to increase at a rapid pace. It is likely that subtle defects in receptor function (such as regulatory or promoter region mutations that increase or decrease receptor gene expression, or mutations in second messenger proteins) will be found that cause endocrine disorders. It is also likely that new receptors will be discovered that transduce or influence hormone action and that endocrine roles will be found for receptors that were not previously thought to mediate or alter hormone action.
References
Related posts:

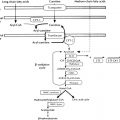
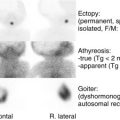
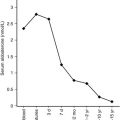
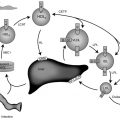

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


