Chapter Outline
GASTROINTESTINAL STROMAL TUMORS
TUMORS OF THE CERVIX AND VAGINA
GESTATIONAL TROPHOBLASTIC TUMOR
This chapter covers the rare tumors of childhood. No attempt has been made to consider the rarest of the rare tumors, and the focus is on the more common of the rare pediatric tumors that do not fall into other categories covered in this text. The chapter is generally organized from top to bottom, with cancers of the head and neck at the beginning and cancers of the gastrointestinal tract discussed later in the chapter. The tumors discussed afflict mostly adults, and some are actually common cancers in older patients. Many are carcinomas and are more familiar to the oncologist who treats adults. These cancers tend to increase in incidence as children reach adolescence.
Evaluation of Surveillance, Epidemiology, and End Results (SEER) data has shown that 9.2% of all childhood cancers are carcinoma, and 75% of these cases are diagnosed in children between the ages of 15 and 19 years. Of the cases in the study, 36% were thyroid carcinomas and 31% were melanomas. The next most common single carcinoma in children younger than age 19 years was nasopharyngeal carcinoma, which comprised 4.5% of these cases. Thus this chapter emphasizes discussion of melanoma, thyroid carcinoma, and nasopharyngeal carcinoma and reviews other rare tumors of childhood.
Oropharyngeal Cancer
Oropharyngeal cancer of squamous cell histology in children remains uncommon. Most of the children who develop epithelial cancer of the nose, ears, and throat are diagnosed with nasopharyngeal carcinoma, which is considered separately in this chapter. The use of smokeless tobacco, cigarette smoking, and alcohol consumption are well established risk factors for the development of squamous cell carcinoma in adults, and the continued use of these substances in Western children warrants monitoring for the development of adult types of squamous cell carcinoma of the oropharynx in children. Although such habits ultimately lead to increased risk for a variety of cancers and early death from heart disease, it remains a theoretical concern that the use of smokeless tobacco by children, particularly by male adolescents, could increase the incidence of these tumors during adolescence and young adulthood.
Duration of smoking increases risk for tobacco-related morbidity and mortality, including several cancers, and significant public health efforts have been aimed at reducing adolescent initiation of tobacco use. Educational interventions have been shown to decrease the prevalence of cigarette and smokeless tobacco use in Massachusetts youth, but restrictions on tobacco sales have a mixed record, because even a complete ban on sales of smokeless tobacco in Finland did not lead to a decrease in its use by adolescents. Conversely, restrictions on smoking in public places have been demonstrated to decrease overall tobacco consumption (e.g., by up to 8% in Italy). Overall trends of cigarette smoking among adolescents have decreased steadily since the mid-1990s. Rates of smoking for white, non-Hispanic twelfth grade students are significantly higher in comparison to Hispanic and African-American students. Although smokeless tobacco use also declined during the late 1990s and early 2000s, recent data from Monitoring the Future and the National Youth Tobacco Survey have demonstrated increased use in recent years, with concurrent decreased reporting of perceived risk. These changes in trends are synchronous with aggressive marketing campaigns by tobacco companies of novel delivery forms of smokeless tobacco such as “snus” and dissolvable tobacco.
The increasing prevalence of human papillomavirus (HPV) infection in adolescents and young adults has raised new concerns about the development of HPV-associated oral squamous-cell carcinoma in younger patients. HPV has recently been detected in approximately 26% of head and neck squamous-cell carcinomas; HPV16 was highly prevalent among these tumors. Trends in adults show an increasing incidence in oral squamous-cell carcinomas despite declining use of tobacco in the United States because of the increasing prevalence of oral HPV infection. A recent study showed 1.7% of 14- to 17-year-old adolescents and 5.6% of 18- to 24-year-old adults had oral HPV; male sex, number of sexual partners, tobacco use, open-mouth kissing, and oral sex have been associated with infection. HPV is most often transmitted in adolescence and young adulthood; a quadrivalent vaccine for HPV that includes strains likely to cause oral cancers is now available and is recommended for both boys and girls.
Few existing studies report outcomes of pediatric squamous-cell carcinomas. A population-based study of 54 pediatric patients with oral-cavity squamous-cell carcinoma had a 5-year disease-specific survival rate of 75%. It is unlikely that many pediatric oncologists will be confronted by adult-type squamous-cell carcinoma of the head and neck, but it is likely that pediatric oncologists will be confronted with tobacco use in young adult and adolescent survivors of pediatric cancer. A St. Jude Children’s Research Hospital study has found that 29% of surviving children treated on an acute myelogenous leukemia (AML) protocol were found to be cigarette smokers at follow-up examinations. Although survivors of childhood cancer may not be seen often in the pediatric oncology clinic, tobacco use and prevention still merit mention during such visits.
Mucoepidermoid carcinoma of the salivary glands is still another type of head and neck carcinoma from which children suffer and is commonly associated with prior therapy for cancer, including radiation. A study of childhood cancer survivors identified 23 cases of secondary head and neck malignancies; of the 14 secondary cancers that arose in the parotid gland, 10 were identified as mucoepidermoid carcinoma. Most children are cured with surgery and sometimes surgery and radiation therapy. Any mass lesion or presenting complaint potentially related to a mass lesion in the salivary glands, particularly if found in a previous radiation field, should prompt a thorough evaluation with suspicion for this tumor.
Nasopharyngeal Carcinoma
Nasopharyngeal carcinoma is a cancer of the epithelial lining of the nasopharynx. It shows varying degrees of differentiation but is a type of squamous-cell carcinoma. Most cases have undifferentiated histology; the undifferentiated type is associated with Epstein-Barr virus (EBV) infection of the nasopharyngeal epithelium, and virus is found in the tumor cells. It is the most common type of epithelial tumor of the head and neck in children, accounting for up to 50% of head and neck tumors in children. Incidence varies widely among populations and in relationship to exposure to EBV, environmental exposures such as preserved or salted fish, and geographic location. Rates in China are particularly high, and ethnic Chinese born in China who move to the West have a higher incidence of the disease than ethnic Chinese who are born in Western countries. In the United States, this cancer is more common in the South and more prevalent in African-American children, who have incidence rates of the disease that approach those of the Chinese. These findings suggest interplay of genetic, viral, and environmental factors in the carcinogenesis of nasopharyngeal carcinoma.
Analysis of incidence and survival rates from SEER data has demonstrated improved survival for Asian-American patients compared to Caucasian and African-American patients. However, nasopharyngeal carcinoma-specific mortality is worse for adults than for children and adolescents, and survival rates in the adolescent and young-adult age ranges tend to be similar among races.
Epidemiology
Nasopharyngeal carcinoma is more common in Asia and is strongly associated with Chinese ethnic origin within Asia, with some parts of China having incidence rates from 15 to 30 cases per 100,000 population. There is also a higher incidence of the cancer in Turkey. A bimodal age distribution has been suggested in nonendemic areas, with an early peak in adolescents age 15 to 19 years. The actual cancer cells are infected by EBV, suggesting a possible role for immunosuppression in the pathogenesis of the tumor, or at least that immune augmentation could help prevent recurrence of nasopharyngeal carcinoma. In Greenland and other regions with similar native populations, high rates of nasopharyngeal carcinoma have been found. Additionally, high rates for other cancers associated with EBV infection, such as various forms of uterine cancer, are more common in families with EBV and nasopharyngeal carcinoma. Population-based screening based on EBV serology in endemic areas can help detect nasopharyngeal carcinoma early. However, this would be impractical in nonendemic areas.
Presentation
The typical presenting signs and symptoms of nasopharyngeal carcinoma include evidence of tumor mass in the nasopharynx such as epistaxis, nasal obstruction and discharge, Eustachian tube dysfunction, palsy of the fifth and sixth nerves relating to skull-based extension, and neck masses. In children with epidemiologic factors making this disease more likely, clinicians must maintain a high index of suspicion for nasopharyngeal carcinoma and obtain imaging studies and consultation as necessary for these signs and symptoms. However, most children diagnosed with nasopharyngeal carcinoma in the United States are unlikely to have obvious epidemiologic associations for the cancer, and even in the United States EBV infection in childhood is common enough that screening based on evidence of EBV infection would be impractical.
Staging and Evaluation
Proper evaluation of patients suspected of having nasopharyngeal carcinoma includes computed tomography (CT) scan and/or magnetic resonance imaging (MRI) of the head and biopsy of appropriate tissue for diagnosis. A clinical examination is key to evaluate for any apparent lymph node spread. Although systemic evaluation, including laboratory testing, positron emission tomography (PET) scanning, whole-body anatomic scanning, and bone marrow biopsy can be considered, they have not been shown to improve staging accuracy in patients with nasopharyngeal carcinoma.
All nasopharyngeal carcinoma cases are considered squamous-cell cancers, but the World Health Organization (WHO) classification of nasopharyngeal carcinoma defines type 1 histology as keratinizing squamous-cell carcinoma, a form of cancer more common in adults, versus the type 2 histology, which does not show keratinization on light microscopy but does show some differentiation. Type 3 histology is nonkeratinized and nondifferentiated and is associated with endemic nasopharyngeal carcinoma. The majority of children with nasopharyngeal carcinoma have a presentation of undifferentiated or WHO type 2 or 3 histology.
The American Joint Commission on Cancer (AJCC) tumor-nodes-metastasis (TNM) staging system (2010) is considered the most relevant for patients in the Western countries. The WHO staging system has fewer local (T) staging categories but seems to be adequate for patients in endemic areas. The latest AJCC staging system is shown in Tables 65-1 and 65-2 .
| Primary Tumor (T) | Regional Lymph Nodes (N) | Distant Metastasis (M) |
|---|---|---|
|
|
|
* The distribution and prognostic impact of regional lymph-node spread from nasopharynx cancer, particularly of the undifferentiated type, are different from those of other head and neck mucosal cancers and justify the use of a different regional lymph-node classification scheme.
† Parapharyngeal extension denotes posterolateral infiltration of tumor.
‡ Midline nodes are considered ipsilateral nodes.
§ Supraclavicular zone or fossa is relevant to the staging of nasopharyngeal carcinoma and is the triangular region originally described by Ho. It is defined by three points: 1) the superior margin of the sternal end of the clavicle; 2) the superior margin of the lateral end of the clavicle; and 3) the point where the neck meets the shoulder. Note that this would include caudal portions of levels IV and VB. All cases with lymph nodes (whole or part) in the fossa are considered N3b.
| Stage | Groupings |
|---|---|
| 0 | Tis, N0, M0 |
| I | T1, N0, M0 |
| II | T1, N1, M0 |
| T2, N0, M0 | |
| T2, N1, M0 | |
| III | T1, N2, M0 |
| T2, N2, M0 | |
| T3, N0, M0 | |
| T3, N1, M0 | |
| T3, N2, M0 | |
| IVA | T4, N0, M0 |
| T4, N1, M0 | |
| T4, N2, M0 | |
| IVB | Any T, N3, M0 |
| IVC | Any T, any N, M1 |
* Results of radiation therapy for nasopharyngeal carcinoma (locoregional control and survival) are usually reported by T stage and N stage separately or by specific T and N subgroupings rather than by numerical stages I to IV. Outcome also depends on various biologic and technical factors related to treatment.
Treatment
Although nasopharyngeal carcinoma can be treated by radiation therapy only, it has been shown to be sensitive to chemotherapy in adults and children. Using chemotherapy may ultimately allow for curative radiotherapy at lower doses with less risk for late effects from head and neck radiotherapy. A Pediatric Oncology Group (POG) study used four cycles of preirradiation chemotherapy with methotrexate with leucovorin, 5-fluorouracil (5-FU), and cisplatin to treat children with AJCC stage III or IV nasopharyngeal carcinoma. Children with stage I or II disease were treated with irradiation only. The 4-year event-free and overall survival rates were 77% ± 12% and 75% ± 12%, respectively. Other agents used to treat nasopharyngeal carcinoma include bleomycin and doxorubicin. More recently, the use of neoadjuvant chemotherapy with concomitant chemoradiation has demonstrated improved outcomes. Xerostomia and dental problems are common adverse effects of treatment. Other long-term morbidities include sensorineural hearing loss, endocrinopathies, trismus, and recurrent infections. Sensorineural hearing loss incidence has been noted to increase with the use of concomitant chemoradiotherapy. Although both children and adults with nasopharyngeal carcinoma are at risk of developing secondary malignancies after treatment, the risk for children is 4.3 times the general population, compared with 1.4 for adults. Current treatment strategies are investigating the use of intensity-modulated radiation therapy (IMRT) and amifostine to preserve salivary function and to decrease acute and chronic sequelae of therapy.
Thyroid Carcinoma
This section summarizes the clinical-pathologic and therapeutic aspects of differentiated thyroid carcinoma in children and adolescents. Medullary thyroid carcinoma (MTC) and associated multiple endocrine neoplasia (MEN) syndromes are not discussed.
Epidemiology
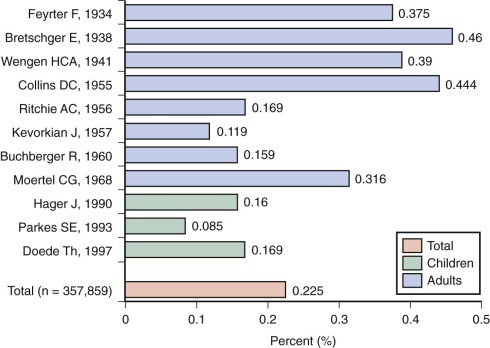
The prevalence of palpable thyroid nodules in children is lower than that seen in adults (1.5% vs. 5%); despite this, thyroid nodules in children are five times more likely to be malignant than those in adults (25% vs. 5%). Of the estimated 56,000 cases of thyroid cancer diagnosed in 2012, only 1.8% occur in patients under 20 years of age. Furthermore, thyroid carcinoma accounts for 3% of all cancers in patients younger than 20 years, and 75% of these occur in patients between the ages of 15 and 19 years ( Fig. 65-1 ). Epidemiological studies suggest that the incidence of thyroid carcinoma has steadily increased over time and that it affects both low and high socioeconomic counties that were surveyed by the SEER registries. In pediatrics, an increasing rate has been documented in the SEER registry for the years 1973 through 2007, with the rates being higher in whites, those aged between 15 and 19 years, girls, and in registries with predominantly white or Hispanic populations.
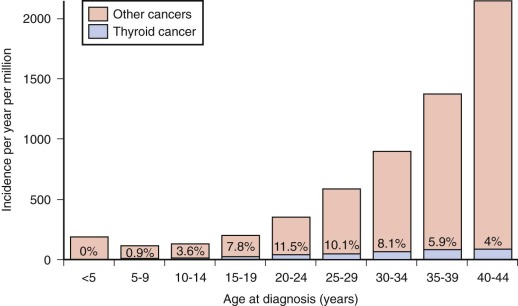
Radioactive exposure to the neck is a well-established risk factor for the development of thyroid carcinoma. Survivors of childhood cancer have up to a 14-fold excess risk of developing thyroid cancer if their thyroid gland received 20 to 25 Gy, and this malignancy accounts for 10% of all subsequent neoplasm in the survivor population. A risk-prediction model for subsequent primary thyroid carcinoma in survivors of childhood cancer has been developed. This model incorporates several variables including radiation and therapy information, age, sex, and a history of a thyroid nodule; the software to compute risk projections can be downloaded at http://dceg.cancer.gov/tools/riskassessment .
Exposure to radioactive iodine and cesium isotopes after the Chernobyl accident resulted in a markedly increased rate of thyroid cancer in children; the estimated relative excess risk was estimated to be 5.6 per Gy of exposure in subjects with estimated doses of less than 1 Gy. Other risk factors associated with the development of pediatric thyroid cancer include various genetic syndromes such as familial adenomatous polyposis (FAP), Cowden disease, Carney syndrome, and immune thyroid disorders such as Hashimoto thyroiditis.
Pathology and Molecular Pathology
More than 90% of pediatric thyroid carcinomas have well-differentiated histology; papillary histology accounts for over 70% of cases, and follicular histology represents about 20% of cases. Medullary thyroid cancer is seen in fewer than 10% of pediatric cases of thyroid cancer and can be found in association with MEN syndrome types 2A and 2B. The distribution of histologies varies slightly in cases with known radiation carcinogenesis; in a study of 740 children with thyroid cancer, of whom 92% were exposed to radiation at Chernobyl, 90% of cases were papillary, about 5% were follicular, and 0.4% were medullary.
RET-PTC rearrangements have been documented in 50% to 60% of pediatric cases of papillary thyroid carcinoma and in up to 70% of radiation-induced pediatric papillary thyroid carcinoma. NTRK1 rearrangements and AKAP9-BRAF fusions have also been described in a small number of radiation-induced papillary tumors. Unlike in adults, BRAF mutations are rarely seen in pediatric papillary tumors. Follicular tumors are characterized by RAS mutations and peroxisome proliferator–activated receptor γ (PPARγ) rearrangements, whereas MEN-associated medullary thyroid carcinomas are characterized by germline-activating mutations of RET.
Clinical Presentation and Staging
Most patients with thyroid carcinoma are older than 10 years and initially have an asymptomatic palpable thyroid nodule. Younger age (<15 years), large fixed nodules, palpable adenopathy, history of radiation exposure, and diseases associated with an increased incidence of thyroid carcinoma such as FAP, Carney complex, Cowden syndrome, MEN types 2A or 2B, pheochromocytoma, and hyperparathyroidism should raise the suspicion for the presence of malignancy.
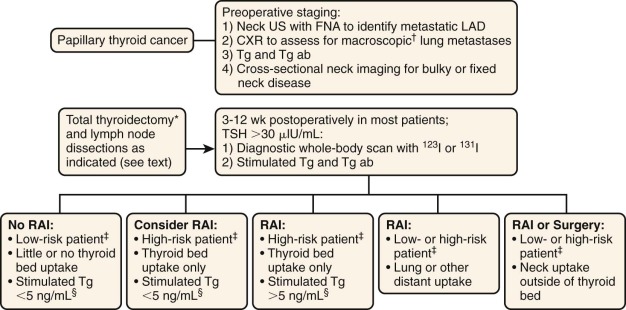
Children and adolescents with thyroid carcinoma are more likely to have nodal regional involvement, extrathyroid disease, and pulmonary metastases and are less likely to die from their disease than adults. Ultrasound is a useful diagnostic technique, and most investigators prefer this method for evaluating thyroid nodules ( Fig. 65-2 ). Initial evaluation of a thyroid nodule should include measurements of serum thyroid-stimulating hormone (TSH), antithyroid peroxidase, antithyroid globulin antibodies, and a neck ultrasound that evaluates the contralateral thyroid lobe and cervical nodes. Screening for MTC using plasma calcitonin may not be cost-effective in children given its low prevalence in the pediatric population. If the TSH level is low, raising the possibility of a hyperfunctional nodule, a thyroid technetium or iodine scan should be performed. Because of the higher likelihood of malignancy in children, it is recommended that all hyperfunctioning nodules be surgically removed in children, with further evaluation and treatment should the pathology be consistent with malignant disease. Fine-needle aspiration biopsy is the most cost-effective and expeditious preoperative procedure to help distinguish benign from malignant thyroid nodules; a meta-analysis of 12 pediatric studies found that this procedure has a sensitivity of 94% and a specificity of 81% (see Fig. 65-2 ). Preoperative staging should include a chest x-ray and a comprehensive neck ultrasound to evaluate the whole thyroid and lymph nodes.
Prognostic Factors
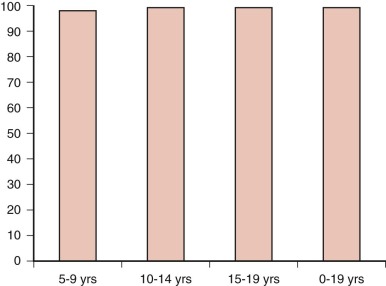
Factors associated with an increased risk of progression include capsular invasion, soft-tissue invasion, positive margins of resection, age younger than 10 to 15 years, male sex, nonpapillary histology, distant metastases, and residual disease after surgery. Survival for children with differentiated thyroid carcinoma approaches 100% ( Fig. 65-3 ).
Treatment and Outcome
Surgery
Surgery is the mainstay of therapy for pediatric and adolescent thyroid carcinoma, and this procedure should be performed by a highly experienced thyroid surgeon. Total thyroidectomy is the initial treatment of choice. especially in younger patients (<10 years) or those with a history of radiation exposure or a family history of thyroid cancer. Many surgeons also advocate the upfront use of this procedure because pediatric thyroid carcinoma is associated with a high incidence of multifocal and metastatic disease, and follow-up screening with serum thyroglobulin is most useful after total thyroidectomy. Lobectomy and isthmectomy can be considered in patients who have a low risk for recurrence such as adolescents with localized small (<1 cm) lesions.
Lymph Node Dissection
Upfront nodal dissection has been shown to decrease the risk of surgical reintervention and nodal recurrence in children with thyroid carcinoma. Current recommendations favor the use of central compartment neck dissection, reserving less extensive surgeries for patients with small tumors and no evidence of nodal involvement by ultrasound (see Fig 65-2 ).
Use of Radioactive Iodine
In adults, the use of radioactive iodine treatment to ablate residual thyroid or iodine-avid metastatic disease decreases the risk of distant metastases. The goals for the use of this therapy in children includes ablation locoregional recurrence, elimination of residual sources of thyroglobulin production, and high remnant uptake that can obscure the presence of metastases and accurate detection of pulmonary metastatic disease. The use of this therapy in children has been associated with a reduced recurrence risk in the thyroid and nodes independent of the surgical approach. However, a recent study by Hay failed to show a significant difference in local and distant recurrence when this modality was used, and 73% of patients who died from a subsequent malignant neoplasm had received therapeutic irradiation postoperatively. Radioactive iodine treatment should be reserved for children at high risk for disease recurrence, and a suggested measured approach for the postoperative use of this agent is shown in Figure 65-2 . When radioactive treatment is prescribed, TSH should be greater than 30 µIU/mL. The dosing of this agent is not well established in children, and various centers follow the American Thyroid Association (ATA) guidelines for adults ( http://thyroidguidelines.net/revised/differentiated ). A posttherapy thyroid scan is usually obtained about 1 week after treatment, and this may identify additional sites of disease that were not apparent initially. Because of the risk of regrowth after TSH exposure, thyroid hormone suppression to achieve serum TSH levels between 0.1 and 0.5 µIU/mL is recommended in all pediatric patients with differentiated thyroid cancer.
Targeted Therapies
A variety of “targeted” therapies have been developed for iodine-refractory differentiated thyroid carcinoma, and many of these agents have been approved for adult use. Most of these therapies target RET and the vascular endothelial growth factor (VEGF) receptors but some also target platelet-derived growth factor β receptors (PDGFRβs), FGFR, MET, and endothelial growth factor (EGF) receptors. Some of these agents include sorafenib, axitinib, motesanib, sunitinib, pazopanib, cabozantinib, and vandetanib. In children sorafenib has been successfully used in a patient with refractory papillary thyroid carcinoma, and in a phase I/II study, vandetanib produced two partial responses in children with MEN2b-associated medullary thyroid carcinoma.
Follow-Up Observation
Lifelong surveillance is recommended for children with thyroid carcinoma. A recent article recommends determination of suppressed thyroglobulin levels and antibodies every 3 to 6 months and a neck ultrasound every 6 to 12 months for the first 2 years of follow-up care. If a patient was treated with radioactive iodine, a stimulated thyroglobulin test and whole body scan is recommended 1 year after initial treatment. About 25% of patients develop antibodies, and these interfere with the thyroglobulin assays. Declining antibody titers usually correlate with lower disease burden.
Breast Tumors
Breast masses are rare in young children. During puberty, benign fibroadenomas are the most common cause of a palpable breast lump ( Table 65-3 ). The estrogen stimulation during adolescence may cause exaggerated growth of the stroma and epithelium in a localized breast lobule that characterizes a fibroadenoma. They have been found in as many as 20% of women between the ages of 15 and 25 years on autopsy studies. Fibroadenoma occurring in adolescence does not appear to be associated with concurrent or future breast carcinoma. Any dominant discrete breast mass in an adolescent should be reexamined at midcycle after one or two menstrual cycles. Persistent masses should be imaged by ultrasound (not mammography) and referred to a surgeon. Typically, malignant breast masses in children have similar imaging characteristics to malignant breast masses in adults, which can help guide the decision to perform more invasive evaluation. Lesions typical for fibroadenoma are sometimes observed clinically; fine-needle aspiration or core biopsy can also be performed to confirm the diagnosis. Most fibroadenomas in adolescents self-resolve or shrink. Most boys with breast masses have gynecomastia that requires only reassurance and noninvasive supportive care. This is borne out by review of pathology specimens from teenage male patients who underwent resection of breast masses; nearly all such evaluations were negative.
| Abnormality | Incidence (%) |
|---|---|
| Fibroadenoma | 68 |
| Miscellaneous | 27 |
| Juvenile hypertrophy | 2 |
| Giant fibroadenoma | 1 |
| Phyllodes tumor | <1 |
| Cancer | <1 |
| Metastatic | 38 |
| Adenocarcinoma (lobular or ductal) | 31 |
| Lymphoma | 13 |
Phyllodes tumors are stromal tumors that present as rapidly growing but clinically benign masses. Although the median age of presentation is 45 years, they can occur in adolescence. Benign phyllodes tumors of the breast are pathologically similar to a large fibroadenoma with more stromal cellularity. They are treated by resection with at least 1 cm margins. Malignant phyllodes tumors are sarcomas with nuclear pleomorphism and mitotic activity; treatment requires a multimodality sarcoma approach. Five-year disease-free survival rates have been reported to be 96%, 74%, and 66% for benign, borderline, and malignant phyllodes tumors, respectively, in adults ; it is thought that adolescents have a more favorable outcome.
Breast masses can also represent primary or metastatic sites of pediatric cancers such as rhabdomyosarcoma, non-Hodgkin lymphoma, Hodgkin lymphoma, or neuroblastoma.
Breast carcinoma in a patient under the age of 20 years is rare; five cases were seen at M.D. Anderson Cancer Center over 10 years, compared with 180 cases in patients between ages 20 and 30 years and 1347 in patients between ages 31 and 50 years. The most common pathology is a juvenile secretory carcinoma that secretes mucin and mucopolysaccharide-containing material. It is treated by excision and lymph node biopsy. No studies have evaluated outcomes in patients younger that the age of 20 years with breast carcinoma, but multiple studies have shown that age younger than 30 or 35 years is an independent predictor of poor response. The two younger age groups at particular risk of breast cancer are women who have received radiation to the breast, usually for lymphoma, and women who have a familial predisposition. These women should be taught breast self-examination and awareness at a young age.
Pulmonary Tumors
Primary tumors of the lung are extremely rare in childhood, and metastatic pulmonary lesions far exceed the number of primary tumors. Table 65-4 lists the most common pulmonary tumors in children.
| Malignant | Benign |
|---|---|
|
|
Bronchial Adenomas
A large percentage of malignant pulmonary tumors in children are called bronchial adenomas, and although not benign, as the term implies, they are predominantly low grade in behavior. They include carcinoid tumor, mucoepidermoid carcinoma, and adenocystic carcinoma. Although pulmonary carcinoid tumors account for only 2% of all primary lung tumors in adults, between 40% and 85% of pediatric malignant lung lesions are carcinoids. See “Carcinoid” for a more complete description of carcinoid and its management. Mucoepidermoid carcinomas (MECs) and adenoid cystic carcinomas are more often seen in the head and neck but can arise from mucous glands located in the respiratory submucosa.
Most bronchial adenomas are endobronchial; patients often have recurrent infections or obstructive pneumonitis before their diagnosis is determined ( Fig. 65-4 ), and the most common presenting symptoms are cough, fever, or wheezing. Although over 25% of adults with pulmonary carcinoid and MEC are asymptomatic, the figure is lower in children, who are less likely to have a radiographic study that would detect an incidental lesion. Aggressive lesions can present with hemoptysis, chest pain, or pneumothorax in rare cases. It is extremely rare for patients with pulmonary carcinoid to show evidence of carcinoid syndrome in the absence of metastatic disease, but a reported 2% to 4% of patients have Cushing syndrome. A mass is usually detected by chest x-ray or chest CT, but endobronchial lesions may only be suspected by recurrent localized obstruction and volume loss ( Fig. 65-5, A and B ). Octreotide scans have a sensitivity of 80% to 90% for carcinoids and can be useful at diagnosis to assess for metastatic disease. More recently, PET CT imaging with the somatostatin analog 68 Ga-DOTATOC has shown sensitivity of 97% and specificity of 92% in identifying neuroendocrine tumors. Diagnostic biopsy is usually obtained by flexible fiberoptic bronchoscopy (see Fig. 65-5, C ). Lymph nodes can be involved, especially with higher grade tumors, but may also be enlarged as a reaction to obstructive pneumonitis rather than tumor.
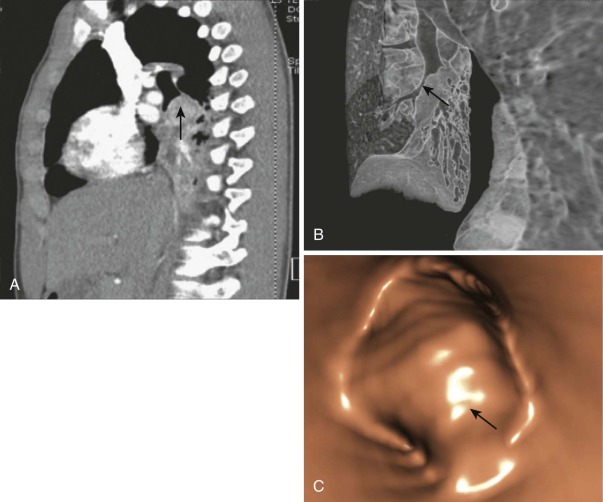
The typical carcinoid tumor and low-grade MEC are pathologically bland and have a low rate of postresection recurrence or metastasis, even with local invasion. The rarer atypical carcinoids and high-grade MEC have a higher mitotic rate and more necrosis and are more likely to behave aggressively. The appropriate therapy is open resection of tumor and involved lymph node, the extent of which is determined by the centrality of the mass. Lobectomy or even pneumonectomy may be required for tumors of the main stem bronchus. Because adenoid cystic carcinomas can spread submucosally with circumferential bronchial involvement, the margin of resection should be determined intraoperatively by frozen sections of the bronchial margins. Local recurrences of bronchial adenomas can occur, especially for MECs, but metastases are rare. Adenoid cystic carcinoma has a greater likelihood of distant metastases than MECs and a poorer overall survival rate. Estimates for metastases from bronchial carcinoid range from 5% to 27%.
When surgery is not curative, other therapies can be used but have shown limited success. Octreotide, a somatostatin analogue used to treat the symptoms of carcinoid syndrome, has been known to stabilize carcinoid tumor growth but not result in regression; similarly, interferon has been used with a modest response. Radionuclide therapy with radiolabelled somatostatin analogs have also been investigated for carcinoids in adults and have demonstrated control of disease and symptom improvement in patients with metastatic disease. Cytotoxic chemotherapy for carcinoid and MECs has been used, but response rates, even with combination agents, are under 20%. Novel molecular agents are being tested, including agents directed against VEGF, PDGFR, and mammalian target of rapamycin (mTor). Prognosis remains excellent for bronchial adenomas, even for locally invasive or high-grade tumors.
Pleuropulmonary Blastoma
Pleuropulmonary blastoma (PPB) in young children is distinct from pulmonary blastoma in adults; the pediatric tumor is composed of primitive blastema and neoplastic mesenchymal cells without epithelial malignant cells. Since its initial description, there has appeared to be an increase in malformations and cancer in the afflicted child and family ( Fig. 65-6 ). Many pleuropulmonary blastomas are now known to be a part of a spectrum of dysplastic and neoplastic lesions that can arise from a constitutional mutation in DICER1, which is located on chromosome 14q and encodes a ribonuclease (RNase) endonuclease essential in the production of micro–ribonucleic acids (miRs). Other lesions that may arise as a result of germline DICER1 mutations include ovarian sex-cord stromal tumors, cystic nephroma, embryonal rhabdomyosarcoma, ciliary body medulloepithelioma, nasal chondromesenchymal hamartoma, and multinodular goiter. The tumor is located peripherally and can be locally invasive. Most patients have symptoms of wheezing, respiratory distress, or pneumothorax. Pathologically there are three subtypes: type 1 PPB, which is exclusively cystic, without a macroscopically detectable solid component); type 2 PPB with solid and true cystic areas; and type 3 PPB, a true solid tumor. PPBs (particularly type 1 cystic lesions) are often misdiagnosed as congenital pulmonary airway malformations (CPAMs) because of similar appearance on chest x-ray and CT imaging. Echocardiography may occasionally demonstrate extension into the thoracic great vessels and heart, and fatal embolic events have been reported in rare occurrences.
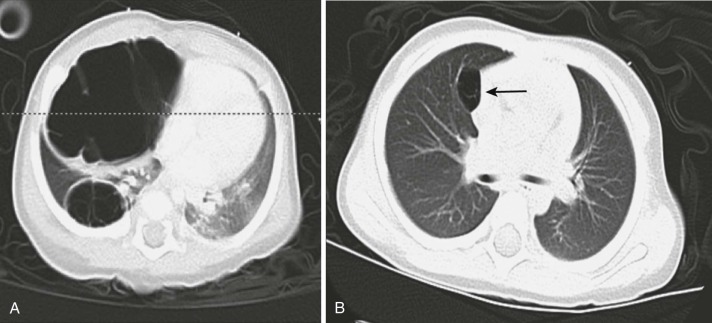
Treatment involves resection. Adjuvant chemotherapy (doxorubicin, actinomycin, vincristine, and cyclophosphamide or ifosfamide), especially for types 2 and 3, may reduce the risk of recurrence or can be used neoadjuvantly to improve the resectability of large lesions. Radiation has been used adjuvantly or palliatively; its role is unclear. If left unresected, type 1 lesions may eventually progress to more aggressive subtypes with development of anaplasia. Prognosis correlates with subtype; type 1 PPB that is completely resected has a 5-year survival rate of 85% to 90%, whereas type 2 and 3 tumors have 45% to 60% survival rates. Metastases can develop in up to 55% of patients with type 3 PPB; the most common sites are the brain and spinal cord and bone. An international PPB registry has been established that collects clinical data on incident cases in an attempt to define the outcomes of patients with this rare disease. Families should be referred for genetic counseling to evaluate for the presence of a DICER1 predisposition syndrome and risk of developing other related lesions.
Bronchogenic Carcinoma
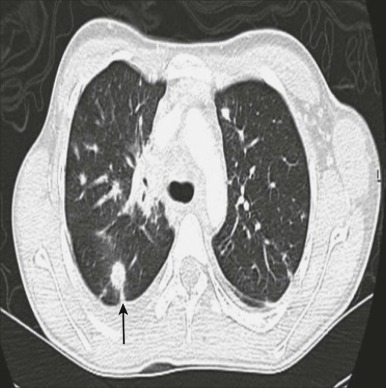
Primary epithelial cancers of the lung are rarely seen in children; fewer than 100 cases have been reported in the literature. They are pathologically indistinguishable from the same tumors occurring in adults. Therapy should be planned after consultation with a medical oncologist experienced with lung cancers and in general should mimic the standard of care for adult tumors. When these histologies occur in children, they are often metastatic at diagnosis and have an aggressive course, high mortality, and average survival time of 7 months after diagnosis ( Fig. 65-7 ). Bronchioalveolar carcinoma (BAC) is a histology that accounts for less than 10% of lung malignancies in adults, with a mean age of presentation between the sixth and seventh decades. In children, mucinous BAC has been associated with CPAM type 1 and has also been reported as a second malignancy in osteosarcoma survivors. Surgery is the primary treatment for BAC. Prognosis depends on nodal status and the resectability of the tumor; it appears to be slightly better for children than for adults with BAC or for children with other primary bronchogenic carcinomas. Squamous cell carcinoma, adenocarcinoma, and small-cell lung cancer are occasionally seen in children. There is no clear causative pathway or association with environmental exposure, and most patients have no family history to suggest a genetic component. A rare form of adenocarcinoma in children is well-differentiated fetal adenocarcinoma (WDFA), which histologically resembles fetal lung and has an excellent prognosis.
Other Pulmonary Tumors
Sarcomas arise from mesenchymal cells and therefore can develop from connective tissue cells in the lung. The most common types seen are hemangiopericytoma, synovial sarcoma, leiomyosarcoma, bronchopulmonary fibrosarcoma, and rhabdomyosarcoma. Ewing sarcoma—peripheral primitive neuroectodermal tumor in the lung (Askin tumor)—is often pleural-based. Given its marked chemosensitivity, it should be treated with chemotherapy before surgical resection in contrast to almost all other lung tumors (see Chapter 61 ). Pleural mesothelioma is occasionally seen in children, with fewer than 100 cases in the literature and even fewer that have been confirmed by pathologic review. Although there have been reports of assumed causation by asbestos exposure and chemotherapy and/or radiation for another malignancy, most cases appear in healthy children. The tumor is similar to that in adult cases in its pathologic appearance and poor prognosis, with a median survival length of approximately 10 months. Most cases are treated with a multimodality approach, but tumors are often diffuse and unresectable and are poorly responsive to chemotherapy and radiation. Current concepts in the therapeutic approach of adults with mesothelioma include the use of cisplatin and multitargeted antifolate chemotherapy and aggressive surgery (extrapleural pneumonectomies).
Gastrointestinal Stromal Tumors
Epidemiology
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasm of the gastrointestinal tract in adults, with an incidence of 11 to 19.6 cases per million, which translates into 3300 to 6000 new cases per year. GISTs are presumed to originate from the interstitial cells of Cajal, which regulate gastrointestinal motility. Bardsley has characterized a putative interstitial cell of Cajal stem cell. This cell is capable of self-renewal and differentiation, has a KIT low CD44+CD34+ phenotype, and is not dependent on KIT signaling for survival and proliferation.
Gastrointestinal Stromal Tumors in Adults
The median age at diagnosis of GIST is approximately 60 years, and the disease has no sex predilection. The most common presenting symptoms are abdominal pain, palpable mass, and gastrointestinal bleeding. In adults, tumors range in size from 1 to 40 cm with a median size of 5 cm and most commonly arise in the stomach (60%) followed by the small intestine (30%) colon, rectum (5%), and esophagus (5%). About 95% of GISTs stain positive for CD117, and activating mutations of KIT (70% to 80%) and PDGFRα (~7%) are seen in nearly 90% of adult GISTs. About 12% to 15% of adult GISTs are wild-type for KIT and PDGFR , but mutations of other genes such as BRAF, SDHB, KRAS, and NRAS have been increasingly recognized ( Table 65-5 ).
| Genetic Type | Frequency of Total | Adjuvant Imatinib | Imatinib for Metastatic Disease |
|---|---|---|---|
| KIT mutated | 80% | ||
| Exon 11 | 67% | Yes, 400 mg/d | Yes, 400 mg/day |
| Exon 9 | 10% | Yes, 400 mg/day | Yes, 800 mg/day |
| Other | 2% | Yes, 400 mg/day | Yes, 400 mg/day |
| PDGFR mutated | |||
| Exon 12 | 5% to 8% | ||
| Exon 14 | 1% | Yes, 400 mg/day | Yes, 400 mg/day |
| Exon 18 | <1% | Yes, 400 mg/day | Yes, 400 mg/day |
| D842V | 5% | No | No |
| Wild-type | 12% to 15% | ||
| BRAF V600 | 7% to 15% | ? | ? |
| SDH A,B,C,D | 2% | ? | ? |
| HRAS/NRAS | <1% | ? | ? |
| Other | 2% | ? | ? |
Chemotherapy for unresectable or metastatic GISTs produces responses in fewer than 10% of patients, and the median survival for adults with metastatic disease is historically 20%. The use of the tyrosine kinase inhibitor imatinib has revolutionized the treatment of GIST. Administration of this drug to adults with unresectable or metastatic GISTs produce responses in up to 85% of patients, and the median survival time for this group has increased to 58 months. The likelihood of response to imatinib is correlated with the mutational genotype of the tumor; patients with KIT exon 11 mutations have significantly higher response rates to imatinib than patients with exon 9 mutations or no mutations. A similar pattern has been documented for patients with PDGFR mutations; two thirds of patients with PDGFR lesions have the imatinib-resistant isoform D842V. Suggested initial management of GISTs based on mutational status is shown in Table 65-5 .
Pediatric Gastrointestinal Stromal Tumors and Succinate Dehydrogenase–Deficient Gastrointestinal Stromal Tumors
Epidemiology and Presenting Features
Pediatric GISTs account for 1% to 2% of all GISTs, but their true incidence in the United States is largely unknown. Review of the SEER database from 2000 through 2009 revealed 23 cases of malignant GIST, and 14 were seen in patients between 15 and 19 years of age for an expected incidence of 0.06 per million among this age group. In children and adolescents these tumors present at a median age of 14 years and predominantly affect females. The most common presenting symptoms are gastrointestinal bleeding, abdominal pain, anemia, weight loss, vomiting and early satiety. About 20% of patients have metastatic disease at diagnosis, and approximately 10% have a history of Carney triad or Carney Stratakis syndrome (see below). GISTs in younger patients more commonly arise in the stomach, tend to be multifocal and involve lymph nodes, more often contain epithelioid or mixed histology, and have a more indolent clinical course. Despite surgical resections, 30% to 80% of patients develop metastatic disease to lymph nodes, liver, or locoregional sites, but despite multiple recurrences, survival is close to 90%.
Biology
In children and adolescents, GISTs can occur within the context of two genetic syndromes: Carney triad and Carney Stratakis syndrome. Carney triad is characterized by pulmonary chondroma, GIST, and paraganglioma, whereas Carney Stratakis syndrome lacks the pulmonary chondromas. Furthermore, Carney triad is sporadic and its cause is not known, whereas Carney Stratakis syndrome is caused by germline inactivating mutations of succinate dehydrogenase (SDH) B, C, or D. Unlike adult GIST in which KIT and PDGFR mutations are key for disease development, only about 15% of GIST tumors found in children have activating mutations of one of these two genes. Despite this their tumors still express KIT at comparable levels to those seen in adults. In contrast to adult GISTs, pediatric tumors lack large-scale chromosomal or genomic aberrations and overexpress IGF1R , suggesting that this pathway should be investigated as a possible therapeutic target in these patients.
About 10% of children with wild-type GIST and no history of paraganglioma have a heterozygous germline mutation of SDH B or C. Additionally, wild-type GIST in patients under 20 years of age almost lack SDHB expression as determined by immunohistochemistry, and nearly 90% of SDHB-negative cases express IGF1R by immunohistochemistry. SDHD germline mutations have been reported in cases of sporadic GIST and in association with familial paraganglioma. Germline SDHA mutations have also been reported in sporadic GIST. Interestingly, about 30% of SDHB-negative cases by immunohistochemistry also lack SDHA expression, and immunohistochemical loss is strongly associated with germline mutations of SDHA. Given the markedly different biology of GIST in younger patients, some investigators prefer using the term SDH-deficient GIST to identify a preferentially younger population that has wild-type GIST and SDH-deficient driven tumors.
Treatment
GIST in younger patients has a more indolent course, and recurrences are common. For this reason, ultimate cure in this group of patients is unlikely and treatment options that combine the judicious use of surgery combined with selected medical therapies offer the best chance for disease control. Because of the rarity of this disease, it is recommended that children and adolescents with GIST be preferentially managed by a multidisciplinary team with expertise in sarcoma and gastrointestinal tumors. Initial management of localized disease should include surgical resection with the goals of achieving a gross resection with an intact pseudocapsule and negative margins. Lymph-node sampling in younger patients should be considered. Gastrectomy and other extensive surgeries should be avoided, because recurrence is common; therefore wedge resections are preferred. The use of adjuvant imatinib in patients with wild-type high-risk tumors is not recommended. For patients with unresectable or metastatic disease who have a KIT or PDGFR mutation, treatment according to Table 65-5 or the National Comprehensive Cancer Network (NCCN) guidelines is recommended. Wild-type or SDH-mutated GISTs do not usually respond to tyrosine kinase therapies used in adults, and in selected asymptomatic patients with active disease, careful observation and follow up care might offer the best approach. In patients who develop significant symptoms or progression, a trial of imatinib or sunitinib is warranted; in one series one partial response and five cases of disease stabilization were seen in children refractory to imatinib. A trial using sunitinib in patients 6 to 21 years if age with wild-type GIST and a trial using the tyrosine kinase inhibitor directed against insulin-like growth factor 1 receptor (IGF1-R) OSI-906 in patients over 18 years with wild-type GIST are ongoing. Follow-up care should include imaging and examinations every 3 to 6 months for 5 years. The use of CT should be limited when possible, and use of MRI and plain chest radiographs encouraged.
Carcinoid Tumors
Carcinoid tumors are tumors of neuroendocrine cells that derive from embryonic divisions of the gut. The most common locations for carcinoid tumors are in locations arising from the midgut: the appendix, small intestine, proximal colon, and rectum. Carcinoid tumor of the appendix is speculated to be the most common tumor of the gastrointestinal tract in children. Tumors of the epithelial component of the bronchial mucosa arising from the foregut appear as carcinoid tumors of the lung and bronchus. Biologically, carcinoid tumor cells contain membrane-bound neurosecretory granules that contain hormones and biogenic amines. When serotonin, 5-hydroxyindole acetic acid (5-HIAA), and other metabolites of the granules are released, a syndrome of flushing, diarrhea, and abdominal cramps can occur. The syndrome rarely occurs in children, because it is associated with large tumor mass, hepatic metastases, or distant metastases. Carcinoid tumors are one of the rarer tumors (less than 10%) seen in the autosomal dominant syndrome MEN type 1 (MEN1), which is caused by a mutation in the tumor suppressor gene MEN1 located at 11q13. MEN1 may be involved in sporadic carcinoid tumors as well, as evidenced by loss of heterozygosity (LOH) on chromosome 11 or inactivated copies of the MEN1 gene. Other possible contributors may be p53, K-ras-2, C-raf-1, and Bax/Bcl2.
In a review of a 22-year period at St. Jude Children’s Research Hospital, 0.08% of patients evaluated for malignancy had carcinoid tumors. In that series, like others, the majority (approximately 60%) arose in the appendix. Pathologic examination of appendectomy specimens reveal carcinoid tumors in 0.23%, with the figure slightly lower in children than adults ( Fig. 65-8 ). Most patients with appendiceal carcinoid tumors have symptoms of acute appendicitis; others have chronic abdominal pain, or an incidental appendectomy is done during another surgical procedure. The diagnosis is rarely made preoperatively.
Related posts:
 The Neonatal Erythrocyte and Its Disorders
The Neonatal Erythrocyte and Its Disorders
 Diagnostic Approach to the Anemic Patient
Diagnostic Approach to the Anemic Patient
 Approach to the Child with a Suspected Bleeding Disorder
Approach to the Child with a Suspected Bleeding Disorder
 Disorders of the Red Cell Membrane
Disorders of the Red Cell Membrane
 Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
 Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


