Abbreviations
- ACTH Adrenocorticotropin
- AZF Azoospermia factor
- cAMP Cyclic adenosine monophosphate
- DHEA Dehydroepiandrosterone
- DHEAS Dehydroepiandrosterone sulfate
- FSH Follicle-stimulating hormone
- GH Growth hormone
- GnRH Gonadotropin-releasing hormone
- hCG Human chorionic gonadotropin
- HESX-1 Hesx-1 homeodomain
- HDL High-density lipoprotein
- HPLC MS/MS High performance liquid chromatography tandem mass spectroscopy
- hGH Human growth hormone
- ICMA Immunochemiluminometric assay
- IGF-1 Insulin-like growth factor 1
- KAL1 Kallmann syndrome
- LDL Low-density lipoprotein
- LH Luteinizing hormone
- LHX3 LIM homeobox gene 3
- PROP-1 Prophet of PIT 1
- PROK1 Prokineticin receptor 2
- PRL Prolactin
- PSA Prostate-specific antigen
- SHBG Sex hormone–binding globulin
- SF-1 Steroidogenic factor 1
- SHOX Short stature homeobox
- SRIF Somatostatin
- TGF-α Transforming growth factor alpha
Puberty: Introduction
Puberty is best considered as one stage in the continuing process of growth and development that begins during gestation and continues until the end of reproductive life. After an interval of childhood quiescence—the juvenile pause—the hypothalamic pulse generator increases activity in the peripubertal period, just before the physical changes of puberty commence. This leads to increased secretion of pituitary gonadotropins and, subsequently, gonadal sex steroids that bring about secondary sexual development, the pubertal growth spurt, and fertility. Historical records show that the age at onset of particular stages of puberty in boys and girls in Western countries has steadily declined over the last several hundred years; this is probably due to improvements in socioeconomic conditions, nutrition, and, therefore, the general state of health during that period. This trend appears to be continuing but now due more to the effects of the obesity epidemic than the improvement in general health.
Many endogenous and exogenous factors can alter age at onset of puberty. While obesity may decrease the age of onset of puberty, chronic illness and malnutrition often delay puberty. There is a significant concordance of age at menarche between mother-daughter pairs and within ethnic populations, indicating the influence of genetic factors. Recent study of genetic loci associated with the age of onset of puberty suggests the existence of several genes that are likely involved in the regulation of menarche and puberty.
Physiology of Puberty
Descriptive standards proposed by Tanner for assessing pubertal development in males and females are in wide use (denoted as Sexual Maturation stages or, often, Tanner stages). They focus attention on specific details of the examination and make it possible to objectively record subtle progression of secondary sexual development that may otherwise be overlooked. Self-assessment of pubertal development by subjects using reference pictures is used in clinical studies but reliability is less than that achieved by physical examination.
The first sign of puberty in the female, as noted in longitudinal studies, is an increase in height velocity that heralds the beginning of the pubertal growth spurt; girls are not usually examined frequently enough to demonstrate this change in clinical practice, so breast development is the first sign of puberty noted by most examiners. Breast development (Figure 15–1) is stimulated chiefly by ovarian estrogen secretion, although other hormones also play a part. The size and shape of the breasts may be determined by genetic and nutritional factors, but the characteristics of the stages in Figure 15–1 are similar in all females. Standards are available for the change in areolar (nipple) plateau diameter during puberty: nipple diameter changes little from stages B1 to B3 (mean of 3-4 mm) but enlarges substantially in subsequent stages (mean of 7.4 mm at stage B4 to 10 mm at stage B5), presumably as a result of increased estrogen secretion at the time of menarche. Areolae become more pigmented and erectile as development progresses. Other features reflecting estrogen action include enlargement of the labia minora and majora, dulling of the vaginal mucosa from its prepubertal reddish hue to more of a pink color (due to cornification of the vaginal epithelium), and production of a clear or slightly whitish vaginal secretion prior to menarche. Pubic hair development (Figure 15–2) is determined chiefly by adrenal and ovarian androgen secretion. Breast development and growth of pubic hair usually proceed at similar rates, but because discrepancies in rates of advancement are possible, it is best to stage breast development separately from pubic hair progression.
Figure 15–1
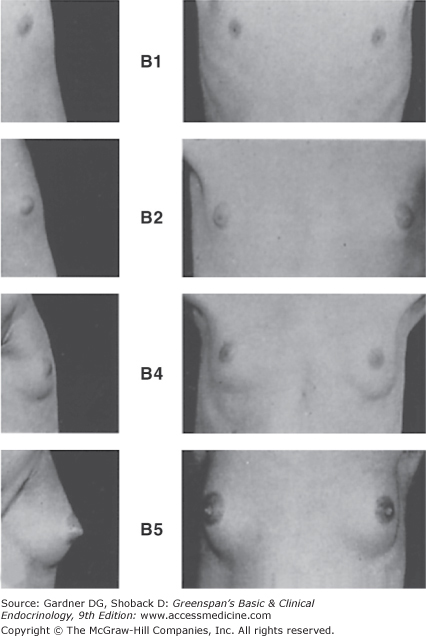
Stages of breast development, according to Marshall and Tanner.
(Photographs from Van Wieringen JC, et al. Growth Diagrams 1965 Netherlands: Second National Survey on0 -24 Year Olds. Institute for Preventive Medicine NO Leiden, Wolters-Noordhoff Publishing; 1971; with permission.)
Stage B1: Preadolescent; elevation of papilla only. Stage B2: Breast bud stage; elevation of breast and papilla as a small mound, and enlargement of areolar diameter. Stage B3 (not shown): Further enlargement of breast and areola with no separation of their contours. Stage B4: Projection of areola and papilla to form a secondary mound above the level of the breast (not shown). Stage B5: Mature stage; projection of papilla only, due to recession of the areola to the general contour of the breast.
Figure 15–2
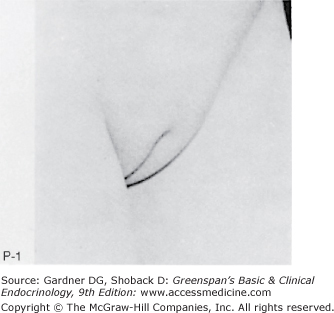
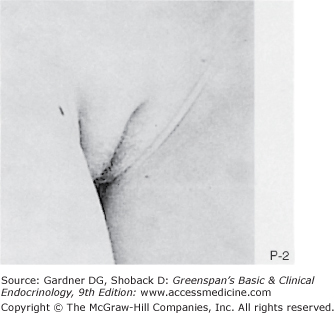
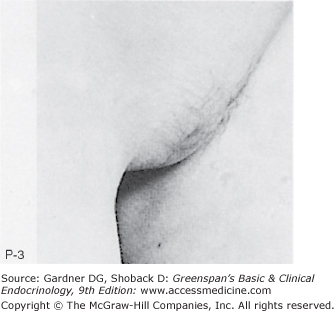
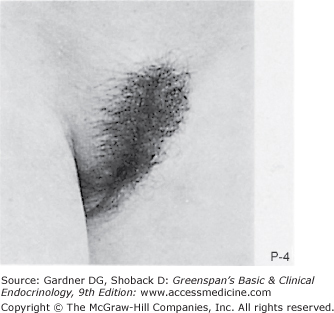
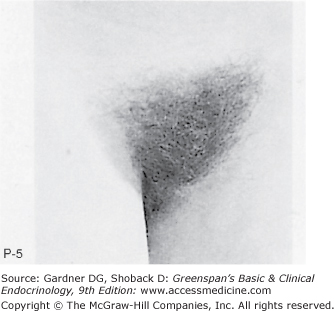
Stages of female pubic hair development, according to Marshall and Tanner.
(Photographs from Van Wieringen JC, et al. Growth Diagrams 1965 Netherlands: Second National Survey on 0 -24 Year Olds. Institute for Preventive Medicine NO Leiden, Wolters-Noordhoff Publishing; 1971; with permission.)
Stage P1: Preadolescent; the vellus over the area is no further developed than that over the anterior abdominal wall (ie, no pubic hair). Stage P2: Sparse growth of long, slightly pigmented, downy hair, straight or only slightly curled, appearing chiefly along the labia. This stage is difficult to see on photographs and is subtle. Stage P3: Hair is considerably darker, coarser, and curlier. The hair spreads sparsely over the superior junction of the labia majora. Stage P4: Hair is now adult in type, but the area covered by it is still considerably smaller than in most adults. There is no spread to the medial surface of the thighs. Stage P5: Hair is adult in quantity and type, distributed as an inverse triangle of the classic feminine pattern. Spread is to the medial surface of the thighs but not up the linea alba or elsewhere above the base of the inverse triangle.
Uterine size and shape change with pubertal development as reflected by ultrasonographic studies. With prolonged estrogen stimulation, the fundus-cervix ratio increases, leading to a bulbous form, and the uterus elongates. An endometrial stripe appears with the onset of puberty that is not found in premature thelarche. Ovaries enlarge with pubertal progression. Small cysts are normally present in prepubertal girls and a multicystic appearance develops during puberty, but the polycystic appearance seen in abnormalities of puberty or during the reproductive years in normal girls is not present. Experienced ultrasonographers can determine the developmental stage of the uterus and ovaries by comparing the results with established standards.
It is important to note that girls achieve reproductive maturity prior to physical maturity and certainly prior to psychologic maturity.
The first sign of normal puberty in boys is usually an increase in the size of the testes to more than 2.5 cm in the longest diameter, excluding the epididymis: this is equivalent to a testicular volume of 4 mL or more. Most of the increase in testicular size is due to seminiferous tubular development secondary to stimulation by follicle-stimulating hormone (FSH), with a smaller component due to Leydig cell stimulation by luteinizing hormone (LH). Thus, if only Leydig cells are stimulated, as with a human chorionic gonadotropin (hCG)-secreting tumor, the testis does not grow as large as in normal puberty. Pubic hair development is brought about by adrenal and testicular androgen secretion and is classified separately from genital development, as noted in Figure 15–3. A longitudinal study of more than 500 boys suggests adding a stage 2a (absence of pubic hair in the presence of a testicular volume of 3 mL or more) to the classic five stages of pubertal development. Further pubertal development occurred in 82% of the subjects in stage 2a after the passage of 6 months: thus, reaching stage 2a would allow the examiner to reassure a patient that further spontaneous development is likely soon. The appearance of spermatozoa in early morning urinary specimens (spermarche) occurs at a mean chronologic age of 13.4 years or a similar mean bone age; this usually occurs at gonadal stage 3 to 4 and pubic hair stage 2 to 4. Remarkably, spermaturia is more common earlier in puberty than later, suggesting that sperm are directly released into the urine early in puberty while ejaculation may be required for the presence of sperm in the urines of older children. However, boys are reported with spermaturia and no secondary sexual development.
Figure 15–3
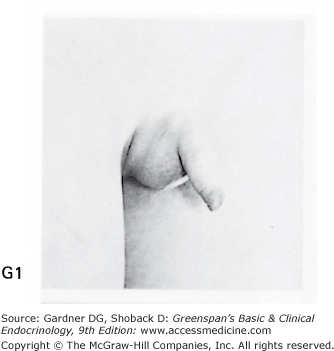
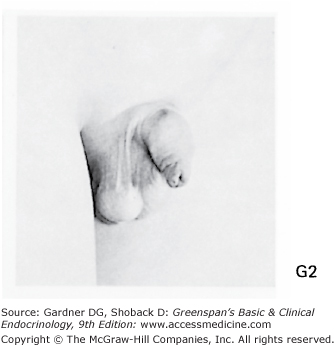
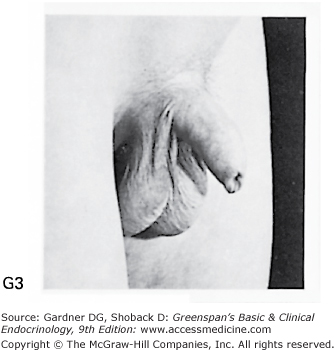
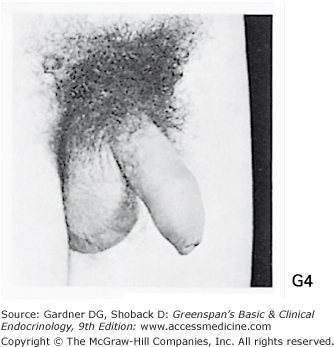
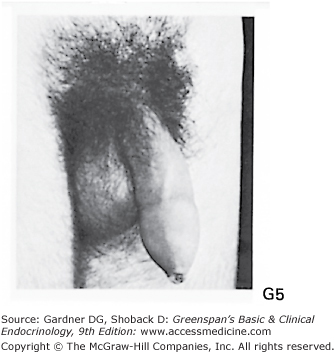
Stages of male genital development and pubic hair development, according to Marshall and Tanner. Genital: Stage G1: Preadolescent. Testes, scrotum, and penis are about the same size and proportion as in early childhood. Stage G2: The scrotum and testes have enlarged, and there is a change in the texture and some reddening of the scrotal skin. There is no enlargement of the penis. Stage G3: Growth of the penis has occurred, at first mainly in length but with some increase in breadth; further growth of testes and scrotum. Stage G4: Penis further enlarged in length and girth with development of glans. Testes and scrotum further enlarged. The scrotal skin has further darkened. Stage G5: Genitalia adult in size and shape. No further enlargement takes place after stage G5 is reached. Pubic hair: Stage P1: Preadolescent. The vellus is no further developed than that over the abdominal wall (ie, no pubic hair). Stage P2: Sparse growth of long, slightly pigmented, downy hair, straight or only slightly curled, appearing chiefly at the base of the penis. This is subtle. Stage P3: Hair is considerably darker, coarser, and curlier and spreads sparsely. Stage P4: Hair is now adult in type, but the area it covers is still considerably smaller than in most adults. There is no spread to the medial surface of the thighs. Stage P5: Hair is adult in quantity and type, distributed as an inverse triangle. Spread is to the medial surface of the thighs but not up the linea alba or elsewhere above the base of the inverse triangle. Most men have further spread of pubic hair.
(Photographs from Van Wieringen JC, et al. Growth Diagrams 1965 Netherlands: Second National Survey on 0-24 Year Olds. Institute for Preventive Medicine NO Leiden, Wolters-Noordhoff Publishing; 1971; with permission.)
It is important to note that boys achieve reproductive maturity prior to physical maturity and certainly prior to psychologic maturity.
Ideally, the upper and lower boundaries encompassing the age at onset of puberty should be set at 2.5 standard deviations (SDs) above and below the mean (encompassing 98.8% of the normal population). Previously, there was no comprehensive study of the start of secondary sexual development adequate to determine the lower limits of normal in US children, as national studies began reporting children who were already 12 years of age or older. Thus, European standards, primarily those of Tanner, were modified for use in the United States. However, a study conducted in medical offices by specially trained pediatricians studying 17,070 girls brought in for routine visits provided information on girls in the United States as young as 3 years. The study revealed that 3% of white girls reach stage 2 breast development by 6 years of age and 5% by 7 years, whereas 6.4% of black girls had stage 2 breast development by 6 years and 15.4% by 7 years. Although this was not a randomly chosen population sampled by longitudinal study (and this led to controversy), it is the largest study available. These data led to the Lawson Wilkins Pediatric Endocrine Society statement that the diagnosis of precocious puberty could be defined as secondary sexual development starting prior to 6 years in black girls and prior to 7 years in white girls who are otherwise healthy. Recent analysis of data from NHANES III demonstrated that children of normal BMI values rarely have breast or pubic hair development before 8 years, although increased BMI in a subset of girls or Hispanic and African American ethnicity led to earlier development. It is essential to use any such guidelines only in healthy girls with absolutely no signs of neurologic or other disease that might pathologically advance puberty or a grave diagnosis may be missed.
Recent data indicate that boys with elevated body mass indices have earlier onset of puberty. However, there has been no change in the age at onset of puberty in boys in the general population, so 9 years is taken as the lower limit of normal pubertal development in males, whereas 13½ years is the upper limit of normal development (although 14 years is often used as a simplified limit). The mean age at menarche in the United States is 12.8 years since the last government study was published in 1974. White girls have menarche later (12.9 years) than black girls (12.3 years), but this 6-month difference is less than the 1-year difference in the age at onset of puberty between the two groups. A compensation occurs in pubertal development so that those girls who enter puberty at the earliest ages of the normal range take more time to reach menarche, and those who enter at older ages of the normal range progress faster to menarche.
There is frequent discussion about an earlier age of puberty in children in the United States, which is demonstrated in some longitudinal studies in the US and abroad. The Bogalusa Heart Study shows that the gap between the age of menarche in Blacks and Whites has widened in the past decades, and this may be due to differing patterns of weight (and fat) gain. However longitudinal study from the Fells Institute shows that those girls who develop early have a tendency to increase their body mass index values after more than those who start puberty later. The presence or absence of menarche however, does not affect the interpretation of a BMI value. Late onset of pubertal development above the upper age limit of normal may indicate hypothalamic, pituitary, or gonadal failure or, alternatively, normal variation (constitutional delay). The time from onset of puberty until adult development is complete is also of importance; significant delays in reaching subsequent stages may indicate any type of hypogonadism.
The striking increase in growth velocity in puberty (pubertal growth spurt) is under complex endocrine control involving thyroid hormone, growth hormone (GH), and estrogen. Hypothyroidism decreases or eliminates the pubertal growth spurt. The amplitude of GH secretion increases in puberty, as does production of insulin-like growth factor (IGF-I); peak serum IGF-I concentrations are reached about 1 year after peak growth velocity, and serum IGF-I levels remain above normal adult levels for up to 4 years thereafter. GH and estrogen are important in the pubertal growth spurt; when either or both are deficient, the growth spurt is decreased or absent. Estrogen indirectly stimulate IGF-I production by increasing the secretion of GH. It also directly stimulates IGF-I production in cartilage. Estrogen is the most important factor in stimulating maturation of the chondrocytes and osteoblasts, ultimately leading to epiphysial fusion. A patient reported with estrogen receptor deficiency was tall, with continued growth past the age of 20 years with a remarkable retardation of skeletal maturation (and decreased bone density). Patients with aromatase deficiency, and therefore, impaired conversion of testosterone to estrogen, also demonstrate diminished advancement of bone age and decreased bone density, as well as continued growth extending into the third decade. With exogenous estrogen administration, the bone age advanced, and the bone density increased. Such patients demonstrate the key role played by estrogen in advancing bone age and bringing about the cessation of growth by epiphysial fusion as well as the importance of estrogen in increasing bone density.
In girls, the pubertal growth spurt begins in early puberty and is mostly completed by menarche. In boys, the pubertal growth spurt occurs toward the end of puberty, at an average age 2 years older than in girls. Total height attained during the growth spurt in girls is about 25 cm; in boys, it is about 28 cm. The mean adult height differential of 12 cm between men and women is due in part to heights already attained before onset of the pubertal growth spurt and in part to the height gained during the spurt.
Twin studies from Finland demonstrate that the heritability estimate (proportion of variance of age of onset of puberty attributable to genetic variance) for age at onset of pubertal growth spurt was 0.91, 0.93 for age at peak height velocity, and 0.97 for adult height. In addition, the age at onset of the pubertal growth spurt was negatively associated with BMI in childhood.
Precocious puberty may coexist with GH deficiency: this situation may occur, for example, in a child with a brain tumor causing central precocious puberty, who is treated with central nervous system radiation that subsequently causes GH deficiency. The precocious puberty may raise the growth rate to the normal range, cloaking the GH deficiency but the child does not grow at the excessive rate characteristic of precocious puberty with GH sufficiency.
Changes in body composition are also prominent during pubertal development. Prepubertal boys and girls start with equal lean body mass, skeletal mass, and body fat, but at maturity men have approximately 1½ times the lean body mass, skeletal mass, and muscle mass of women, whereas women have twice as much body fat as men. Attainment of peak values of percentage of body fat, lean body mass, and bone mineral density is earlier by several years in girls than in boys, as is the earlier peak of height velocity and velocity of weight gain in girls.
The most important phases of bone accretion occur during infancy and during puberty. Girls reach peak mineralization between 14 and 16 years of age, whereas boys reach a later peak at 17.5 years; both milestones occur after peak height velocity (Figure 15–4). The density of bone is determined by genetic factors; decreased bone mass is found in familial patterns even if subjects are studied before puberty. Patients with delay in puberty for any reason have a significant decrease in bone accretion and a delay in reaching peak bone mineral density, although bone density may later reach normal values in those with constitutional delay in puberty. Moderate exercise increases bone mass, but excessive exercise itself delays puberty; the ultimate outcome of excessive exercise in girls is the combination of exercise-induced amenorrhea, premature osteoporosis, and disordered eating, which is known as the female athletic triad.
Figure 15–4
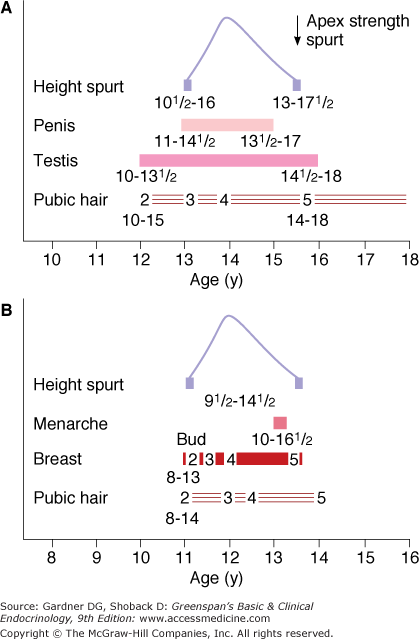
Sequence of secondary sexual development in British males (A) and females (B). The range of ages in Britain is indicated. American boys and girls start pubertal stages earlier than British children (see text) but the sequence is the same.
(Reproduced, with permission, from Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. 1970;45:13.)
Unfortunately, in the United States, only a minority of adolescents receive the recommended daily allowance of calcium (>1000 mg/d depending on age) and vitamin D, and a future epidemic of osteopenia or even osteoporosis in normal subjects who have this deficiency is a possibility although genetic influences are prominent in later adult bone density. It is especially important to ensure adequate calcium intake and vitamin D in patients with delayed or absent puberty and in patients treated with gonadotropin-releasing hormone (GnRH) agonists.
Other changes that are characteristic of puberty are mediated either directly or indirectly by the change in sex steroids. For example, seborrheic dermatitis may appear, the mouth flora changes, and periodontal disease, rare in childhood, may occur. Insulin resistance intensifies in normal adolescents as well as those with type 1 diabetes mellitus.
Pituitary gonadotropin secretion is controlled by the hypothalamus, which releases pulses of GnRH into the pituitary-portal system to reach the anterior pituitary gland by 20 weeks of gestation. Control of GnRH secretion is exerted by a hypothalamic pulse generator in the arcuate nucleus. It is sensitive to feedback control from sex steroids and inhibin, a gonadal protein product that controls the frequency and amplitude of gonadotropin secretion during development in both sexes and during the progression of the menstrual cycle in females (see Chapter 13).
In males, LH stimulates the Leydig cells to secrete testosterone, and FSH stimulates the Sertoli cells to produce inhibin. Inhibin feeds back on the hypothalamic-pituitary axis to inhibit FSH. Inhibin is also released in a pulsatile pattern, but concentrations do not change with pubertal progression.
In females, FSH stimulates the granulosa cells to produce estrogen and the follicles to secrete inhibin, and LH appears to play a minor role in the endocrine milieu until menarche. Then LH triggers ovulation and later stimulates the theca cells to secrete androgens (see Chapter 13).
The concept of the continuum of development between the fetus and the adult is well illustrated by the changes that occur in the hypothalamic-pituitary-gonadal axis. Gonadotropins are demonstrable in fetal pituitary glands and serum during the first trimester. The pituitary content of gonadotropins rises to a plateau at midgestation. Fetal serum concentrations of LH and FSH rise to a peak at midgestation and then gradually decrease until term. During the first half of gestation, hypothalamic GnRH content also increases, and the hypophyseal-portal circulation achieves anatomic maturity. These data are compatible with a theory of early unrestrained GnRH secretion stimulating pituitary gonadotropin secretion, followed by the appearance of central nervous system factors that inhibit GnRH release and decrease gonadotropin secretion after midgestation. Because the male fetus has measurable serum testosterone concentrations but lower serum gonadotropin concentrations than the female fetus, negative feedback inhibition of gonadotropin secretion by testosterone appears to operate after midgestation.
At term, serum gonadotropin concentrations are suppressed in the neonate, but with postnatal clearance of high circulating estrogen concentrations, negative inhibition is reduced and postnatal peaks of serum LH and FSH are measurable for several months to up to a few years after birth. Serum testosterone concentrations may be increased to midpubertal levels during the several months after birth in normal males, and serum estradiol rises in pulses for the first 2 years in infant girls. These peaks of gonadotropins and sex steroids in normal infants complicate the diagnosis of central precocious puberty at these youngest ages, because it is difficult to decide whether to attribute the gonadotropin and sex steroid peaks to central precocious puberty or to normal physiology (the mini-puberty of infancy). While episodic peaks of serum gonadotropins may occur until 2 years of age, serum gonadotropin concentrations are low during later years in normal childhood.
While serum gonadotropin concentrations are low in midchildhood, sensitive assays indicate that pulsatile secretion occurs and that the onset of puberty is heralded more by an increase in amplitude of secretory events than a change in frequency. Twenty-four-hour mean concentrations of LH, FSH, and testosterone rise measurably within 1 year before the development of physical pubertal changes. Patients with gonadal failure—such as those with the syndrome of gonadal dysgenesis (Turner syndrome)—demonstrate an exaggeration of the normal pattern of gonadotropin secretion, with exceedingly high concentrations of serum LH and FSH during the first several years of life. Such patients show that negative feedback inhibition is active during childhood; without sex steroid or inhibin secretion to exert inhibition, serum gonadotropin values are greatly elevated. During midchildhood, normal individuals and patients with primary hypogonadism have lower serum gonadotropin levels than they do in the neonatal period, but the range of serum gonadotropin concentrations in hypogonadal patients during midchildhood is still higher than that found in healthy children of the same age. The decrease in serum gonadotropin concentrations in primary agonadal children during midchildhood is incompletely understood, but has been attributed to an increase in the central nervous system inhibition of gonadotropin secretion during these years. Thus, the juvenile pause in normal children and those with primary gonadal failure appears to be due to central nervous system restraint of GnRH secretion.
Prepubertal children demonstrate a circadian rhythm of LH and FSH secretion of low amplitude with a pattern of low levels of sex steroid secretion lagging behind the gonadotropin rhythm. The delay is presumably due to the time necessary for biosynthesis and secretion of sex steroids. Thus, the changes that are described later and that occur at puberty do not arise de novo but are based on preexisting patterns of endocrine secretion. In the peripubertal period, endogenous GnRH secretion increases in amplitude and frequency during the early hours of sleep and serum testosterone and estrogen concentrations rise several hours later, suggesting that biosynthesis or aromatization occurs during the period of delay—a pattern that differs from the prepubertal period mainly in the increased amplitude of the secretion encountered in puberty. As puberty progresses in both sexes, the peaks of serum LH and FSH occur more often during waking hours; and, finally, in late puberty, the peaks occur at all times, eliminating the diurnal variation.
During the peripubertal period of endocrine change prior to secondary sexual development, gonadotropin secretion becomes less sensitive to negative feedback inhibition. Before this time, a small dose of exogenous sex steroids virtually eliminates gonadotropin secretion, whereas afterward a far larger dose is required to suppress serum FSH and LH. Thus, an equilibrium of pubertal concentrations of gonadotropins and sex steroids is reached.
The switch that triggers the onset of puberty is unknown, but several neurotransmitters are invoked in the process, including gamma-aminobutyric acid and N-methyl-d-aspartate. Most recently, KISS1, a human metastasis suppressor gene at locus 19p13.3, which codes for metastin (or kisspeptin), a 145 amino acid peptide, has been implicated. Metastin is the endogenous agonist for GPR54, a Gq/11-coupled receptor of the rhodopsin family (metastin receptor), which is found in the brain, mainly in the hypothalamus and basal ganglia, as well as in the placenta. KISS1 mRNA levels increase with puberty in intact male and female monkeys from the juvenile to midpubertal stages. Furthermore, administration of KISS1 via intracerebral catheters to GnRH-primed juvenile female rhesus monkeys stimulates GnRH release, release that is abolished by infusion of GnRH antagonist. Thus, it is postulated that KISS1 signaling through the GPR54 receptor of primate hypothalamus may be activated at the end of the juvenile pause and contribute to the pubertal resurgence of pulsatile GnRH release of puberty, serving as a trigger for the onset of puberty.
Highly sensitive sandwich assays (immunoradiometric [IRMA]) and immunochemiluminometric assays (ICMA) are available for gonadotropin determination. They can be used to indicate the onset of pubertal development solely with basal samples usually without the necessity for GnRH testing (see GnRH and GnRH agonist testing in the Central Precocious Puberty section). Elevated basal LH values (>0.3 IU/L) determined by third-generation assays in random blood samples are highly predictive of elevated peak GnRH-stimulated LH and therefore indicate the onset of central precocious puberty or normal puberty. These third-generation assays further reflect the remarkable logarithmic increase in spontaneous LH secretion in the latest stages of prepuberty and earliest stages of puberty as the testicular volume increases from 1 mL to 10 mL; these increases in serum LH are far greater proportionately than those found in the last stages of pubertal development. The magnitude of increase in serum testosterone is also greater in the early stages of puberty and correlates with the increase in serum LH during this same period of early pubertal development. Note that standard LH and FSH assays are not sensitive enough to detect the changes seen with the ultrasensitive assays. Furthermore, the laboratory must have pubertal standard values available to permit accurate interpretation of the measurements. Since both types of assays are available commercially, care in ordering is needed.
Sex steroid secretion is correlated with the development of gonadotropin secretion. During the postnatal period of increased episodic gonadotropin secretion, plasma concentrations of gonadal steroids are episodically elevated. This indicates the potential for secretory activity in the neonatal gonad. Later, when gonadotropin secretion decreases in midchildhood, gonadal activity decreases, but the testes can still be stimulated by LH or hCG and the ovaries by FSH, with resulting secretion of gonadal steroids. An ultrasensitive estradiol assay demonstrates higher values of serum estradiol in prepubertal girls than prepubertal boys, indicating definite basal ovarian activity during the juvenile pause. With the onset of puberty, serum gonadal steroid concentrations progressively increase. While sex steroids are secreted in a diurnal rhythm in early puberty, as are gonadotropins, sex steroids are bound to sex hormone–binding globulin (SHBG), so that the half-life of sex steroids is longer than that of gonadotropins. Thus, random daytime measurements of serum sex steroids are more helpful in determining pubertal status than random measurements of serum gonadotropins but still are not infallible.
Most (97%-99%) of the circulating estradiol and testosterone is associated with SHBG. The free hormone is the active fraction, but SHBG modulates the activity of the total testosterone and estradiol. Prepubertal boys and girls have equal concentrations of SHBG, but because testosterone decreases SHBG and estrogen increases SHBG, adult males have only half the concentration of SHBG compared with adult females. Thus, lower SHBG levels amplify androgen effects in men. While adult men have 20 times the amount of plasma testosterone that adult women have, adult men have 40 times the amount of free testosterone that adult women have.
The use of intravenous, exogenous GnRH has further clarified the pattern of pubertal development. When GnRH is administered to children less than 2 years of age, pituitary secretion of LH and FSH increases markedly. During the juvenile pause, the period of low basal gonadotropin secretion (after age 2 until the peripubertal period), exogenous GnRH has less effect on LH release. By the peripubertal period, intravenous GnRH or GnRH agonist induces a greater rise in LH concentrations in boys and girls, and this response continues until adulthood. There is no significant change in the magnitude of FSH secretion after GnRH with the onset of puberty, though females at all ages release more FSH than males.
Gonadotropins are released in secretory spurts in response to endogenous GnRH, which itself is secreted episodically about every 90 to 120 minutes in response to a central nervous system pulse generator. GnRH can be administered to patients in episodic boluses by a programmable pump that mimics the natural secretory episodes. A prepubertal subject without significant gonadotropin peaks demonstrates the normal pubertal pattern of episodic secretion of gonadotropins after only a few days of exogenously administered GnRH boluses. Hypogonadotropic patients, who in the basal state do not have normal secretory episodes of gonadotropin release, may be converted to a pattern of normal adult episodic gonadotropin secretion by this method of pulsatile GnRH administration.
This phenomenon is used in clinical practice to bring about ovulation or spermatogenesis to foster fertility. Varying the timing of pulsatile GnRH administration can regulate the ratio of FSH to LH just as the frequency of endogenous hypothalamic GnRH release shifts during the menstrual cycle and puberty to alter this ratio naturally. Increasing the frequency of GnRH pulses increases the LH-FSH ratio; an increased ratio is characteristic of midcycle and peripubertal dynamics. Alternatively, if GnRH is administered continuously rather than in pulses or if long-acting superactive analogs of GnRH are given, a brief period of increased gonadotropin secretion is followed by LH and FSH suppression (see later). This phenomenon is responsible for the therapeutic effects of GnRH analogs in conditions such as central precocious puberty.
Leptin is a hormone produced in adipose cells that suppresses appetite through interaction with its receptor in the hypothalamus. Leptin plays a major role in pubertal development in mice and rats. Genetically leptin-deficient mice (ob/ob) do not initiate puberty. Leptin replacement promotes pubertal development in this mouse, and leptin administration causes an immature normal mouse to progress through puberty. A leptin-deficient girl at 9 years of age was remarkably obese and had a bone age of 13 years (usually appropriate for the onset of normal puberty) but no significant gonadotropin pulses and no physical evidence of pubertal development. With leptin treatment, gonadotropin peaks appeared, and pubertal development followed. Individuals with leptin receptor deficiency also have disordered puberty. While these and other data suggested that leptin might be the elusive factor that triggers the onset of puberty, clinical data proved otherwise. Longitudinal studies demonstrate that leptin increases in girls during puberty in synchrony with the increase in fat mass, while leptin decreases in boys, with increased lean body mass and decreased fat mass in relation to testosterone production. While leptin does not appear to trigger the onset of puberty in normal adolescents, leptin may accompany pubertal changes rather than cause them. Leptin is a necessary component of pubertal development in human beings but not a major stimulant to this development.
The last stage in hypothalamic-pituitary development is the onset of positive feedback, leading to ovulation and menarche. The ovary contains a paracrine system that regulates follicular atresia or development; it is only in the last stages of puberty that gonadotropins come into play in the maturation of the follicle. After midpuberty, estrogen in the appropriate amount at the appropriate time can stimulate gonadotropin release, whereas higher doses of estrogen can suppress gonadotropin secretion. The frequency of pulsatile GnRH release increases during the late follicular phase of the normal menstrual cycle and raises the ratio of LH to FSH secretion. This stimulates the ovary to produce more estrogen and leads to the midcycle LH surge that causes ovulation. Administration of pulsatile GnRH by programmable pump can be used to bring about fertility in patients with hypothalamic GnRH deficiency by mimicking this natural pattern as noted earlier.
However, even if the midcycle surge of gonadotropins is present, ovulation may not occur during the first menstrual cycles; 90% of menstrual cycles are anovulatory in the first year after menarche, and it is not until 4 to 5 years after menarche that the percentage of anovulatory cycles decreases to less than 20%. This high prevalence of anovulatory periods may be due to unrecognized PCOS in the young rather than a normal developmental phenomenon in some individuals. However, some of the first cycles after menarche may be ovulatory, and fertility is possible in the first cycle.
Just as boys are reproductively mature prior to physical maturity, girls may become fertile and even pregnant prior to physical or emotional maturity.
Although the hypothalamic-pituitary axis has been well characterized in recent years, our understanding of the mechanism of control of adrenal androgen secretion is still somewhat rudimentary. The adrenal cortex normally secretes the weak androgens dehydroepiandrosterone (DHEA), its sulfate, dehydroepiandrosterone sulfate (DHEAS), and androstenedione in increasing amounts beginning at about 6 to 7 years of age in girls and 7 to 8 years of age in boys (Table 15–1). A continued rise in adrenal androgen secretion persists until late puberty. Thus, adrenarche (the secretion of adrenal androgens) occurs years before gonadarche (the secretion of gonadal sex steroids). The observation that patients with Addison disease, who do not secrete adrenal androgens, and patients with premature adrenarche, who secrete increased amounts of adrenal androgens at an early age, usually enter gonadarche at a normal age. This suggests that age at adrenarche does not significantly influence age at gonadarche. Furthermore, patients treated with a GnRH agonist to suppress gonadotropin secretion progress through adrenarche despite their suppressed gonadarche.
| Boys | Girls | |||||||
|---|---|---|---|---|---|---|---|---|
| Tanner Stage | LH (IU/L) | FSH (IU/L) | Testosterone (ng/d L) | DHEAS (μg/d L) | LH (IU/L) | FSH (IU/L) | Estradiol (pg/mL) | DHEAS (μg/dL) |
| I | ≤0.52 | ≤3.0 | ≤5 | ≤89 | ≤0.15 | 0.50-4.50 | ≤16 | ≤46 |
| II | ≤1.76 | 0.30-4.00 | ≤167 | ≤81 | ≤2.91 | 0.40-6.50 | ≤65 | 15-113 |
| III | ≤4.06 | 0.30-4.00 | 21-719 | 22-126 | ≤7.01 | 0.40-6.50 | ≤142 | 42-162 |
| IV | 0.06-4.77 | 0.40-7.40 | 25-912 | 33-177 | 0.10-14.7 | 0.80-8.50 | ≤283 | 42-241 |
| V | 0.06-4.77 | 0.40-7.40 | 110-975 | 110-510 | 0.10-14.7 | 0.80-8.50 | aSee below | 45-320 |
The onset of puberty is associated with many changes in laboratory values that are either directly or indirectly caused by the rise of sex steroid concentrations. Thus, in boys, hematocrit rises, and high-density lipoprotein (HDL) concentrations fall as a consequence of increasing testosterone. In both boys and girls, alkaline phosphatase rises normally during the pubertal growth spurt (often this rise is incorrectly interpreted as evidence for a tumor or liver abnormality). Serum IGF-I concentrations rise with the growth spurt, but IGF-I is more closely correlated with sex steroid concentration than with growth rate. IGF-I levels peak 1 year after peak growth velocity is reached and remain elevated for 4 years thereafter, even though growth rate is decreasing. Prostate-specific antigen (PSA) is measurable after the onset of puberty in boys and provides another biochemical indication of pubertal onset.
Delayed Puberty or Absent Puberty (Sexual Infantilism)
Any girl of 13 or boy of 14 years of age without signs of pubertal development falls more than 2.5 SD above the mean and is considered to have delayed puberty (Table 15–2). By this definition, 0.6% of the healthy population is classified as having delay in growth and adolescence. These normal patients need reassurance rather than treatment and ultimately progress through the normal stages of puberty, albeit later than their peers. The examining physician must make the sometimes difficult decision about which patients older than these guidelines are constitutionally delayed and which have organic disease.
| Constitutional delay in growth and adolescence |
| Hypogonadotropic hypogonadism |
| Central nervous system disorders |
| Congenital disorders of the hypothalamus or pituitary |
| Tumors |
| Other acquired disorders |
| Infection |
| Trauma |
| Irradiation |
| Defects of the hypothalamic-pituitary axis |
| Isolated gonadotropin deficiency |
| Kallmann syndrome |
| Gonadotropin deficiency with normal sense of smell |
| Multiple pituitary hormonal deficiencies |
| Miscellaneous disorders |
| Prader-Willi syndrome |
| Laurence-Moon, Bardet-Biedl syndromes |
| Chronic disease |
| Weight loss |
| Anorexia nervosa |
| Increased physical activity in female athletes |
| Hypothyroidism |
| Hypergonadotropic hypogonadism |
| Male phenotype |
| Klinefelter syndrome |
| Other forms of primary testicular failure (including chemotherapy) |
| Enzymatic defects of androgen production |
| Anorchia or cryptorchism |
| Female phenotype |
| Turner syndrome |
| Other forms of primary ovarian failure (including chemotherapy |
| Pseudo–Turner syndrome |
| Noonan syndrome |
| XX and XY gonadal dysgenesis |
A patient with delayed onset of secondary sexual development, whose stature is shorter than that of age-matched peers but who consistently maintains a normal growth velocity for bone age and whose skeletal development is delayed more than 2 SD from the mean, is likely to have constitutional delay in puberty (Figure 15–5). These patients are at the older end of the normal distribution curve describing the age at onset of puberty. A family history of a similar pattern of development in a parent or sibling supports the diagnosis. The subject is usually thin as well. In many cases, even if they show no physical signs of puberty at the time of examination, the initial elevation of gonadal sex steroids has already begun, and their basal LH concentrations measured by ultrasensitive third-generation assays or their plasma LH response to intravenous GnRH or GnRH agonist is pubertal. In boys, an 8 am serum testosterone value above 20 ng/dL (0.7 mmol/L) also indicates that secondary sexual development will commence within a period of months.
Figure 15–5


Girl, 13-4/12 years old, with constitutional delay in growth and puberty. History revealed a normal growth rate but short stature at all ages. Physical examination revealed a height of 138 cm (−4.5 SD) and a weight of 28.6 kg (−3 SD). The patient recently had early stage 2 breast development, with 1 cm of glandular tissue on the right breast and 2 cm on the left breast. The vaginal mucosa was dulled, and there was no pubic hair. Karyotype was 46,XX. Bone age was 10 years. After administration of GnRH, LH, and FSH rose in a pubertal pattern. Estradiol was 40 pg/mL. She has since spontaneously progressed through further pubertal development.
(Reproduced, with permission, from Styne DM, Kaplan SL. Pediatr Clin North Am. 1979;26:123.)
In some cases, observation for endocrine or physical signs of puberty must continue for a period of months or years before the diagnosis is made. Generally, signs of puberty appear after the patient reaches a skeletal age of 11 years (girls) or 12 years (boys), but there is great variation. Patients with constitutional delay in adolescence almost always manifest secondary sexual development by 18 years of chronologic age, although there is one reported case of spontaneous puberty occurring at 25 years of age. Reports of patients with Kallmann syndrome and others with constitutional delay in puberty within one family suggest a possible relationship between the two conditions (see later and Chapter 4). Adrenarche is characteristically delayed—along with gonadarche—in constitutional delay in puberty.
The absent or decreased ability of the hypothalamus to secrete GnRH or of the pituitary gland to secrete LH and FSH leads to hypogonadotropic hypogonadism. This classification suggests an irreversible condition requiring replacement therapy. If the pituitary deficiency is limited to gonadotropins, patients are usually close to normal height for age until the age of the pubertal growth spurt, in contrast to the shorter patients with constitutional delay. Bone age is usually not delayed in childhood but does not progress normally after the patient reaches the age at which sex steroid secretion ordinarily stimulates maturation of the skeleton. Patients may reach taller stature than expected. However, if GH deficiency accompanies gonadotropin deficiency, severe short stature will result.