Abbreviations
- ACTH Adrenocorticotropic hormone
- CT Computed tomography
- GDNF Glial cell line-derived neurotrophic factor
- GDNFR Glial cell line-derived neurotrophic factor receptor
- GNAS Gene encoding the alpha subunit of a stimulatory G protein
- MCT Medullary carcinoma of thyroid
- MEN Multiple endocrine neoplasia
- MRI Magnetic resonance imaging
- PCR Polymerase chain reaction
- PTH Parathyroid hormone
- RET Rearranged during transfection proto-oncogene
- VIP Vasoactive intestinal peptide
- ZES Zollinger-Ellison syndrome
Multiple Endocrine Neoplasia: Introduction
A group of heritable syndromes characterized by aberrant growth of benign or malignant tumors in a subset of endocrine tissues have been given the collective term multiple endocrine neoplasia (MEN). The tumors may be functional (ie, capable of elaborating hormonal products that result in specific clinical findings characteristic of the hormone excess state) or nonfunctional. There are three major syndromes: MEN1 is characterized by tumors involving the parathyroid glands, the endocrine pancreas, and the pituitary; MEN 2A includes medullary carcinoma of the thyroid (MCT), pheochromocytoma, and hyperparathyroidism; and MEN 2B, like MEN 2A, includes MCT and pheochromocytoma, but hyperparathyroidism is typically absent.
Multiple Endocrine Neoplasia Type 1
MEN1, also known as Wermer syndrome, is inherited as an autosomal dominant trait with an estimated prevalence of 2 to 20 per 100,000 in the general population. Approximately 10% of MEN1 mutations arise de novo. The term sporadic MEN1 has been applied to this group. MEN1 has a number of unusual clinical manifestations (Table 22–1) that occur with variable frequency among individuals within affected kindreds. The earliest age at which manifestations of MEN1 have been reported is 5 years; penetrance is maximal by the fifth decade of life.
Hyperparathyroidism is the most common feature of MEN1, with an estimated penetrance of 95% to 100% over the lifetime of an individual harboring the MEN1 gene. The diagnosis of hyperparathyroidism is usually made through a combination of clinical and laboratory criteria similar to those used in the identification of sporadic disease (see Chapter 8). It is typically the first clinical manifestation of MEN1, although this varies as a function of the patient population being examined. Hyperparathyroidism in MEN1 is due to hyperplasia of all four parathyroid glands (or more, if supernumerary glands are present). However, involved glands may undergo metachronous enlargement, and selective resection of these glands often results in sustained clinical remissions. MEN1 is a rare cause of hyperparathyroidism, accounting for only 2% to 4% of cases in the general population.
Enteropancreatic tumors in MEN1 (∼50% of all MEN1 patients) can be either functional (ie, capable of producing a secreted product with biologic activity) or nonfunctional. Gastrinomas, frequently associated with Zollinger-Ellison syndrome (ZES), represent approximately 40% to 60% of the enteropancreatic tumors associated with this syndrome. Of equal importance, roughly 25% of patients with ZES are found in MEN1 kindreds. Insulinomas constitute approximately 20% of the islet cell tumors, and the remainder represent a collection of functional (eg, glucagon- or vasoactive intestinal peptide [VIP]-producing tumors) and nonfunctional tumors. It is noteworthy that the gastrinomas of MEN1 are often small, multicentric, and ectopically located outside the pancreatic bed, most often in the duodenal submucosa. This latter feature can have a major impact on the therapeutic approach to these patients (discussed later). Gastrinomas in MEN1 are frequently malignant, as are their sporadic counterparts; however, for reasons that are only poorly understood, the biologic behavior of these tumors is less aggressive than those found in sporadic diseases.
The diagnosis of gastrinomas is based on demonstration of hypergastrinemia in the presence of gastric acid hypersecretion. This latter criterion excludes other more common causes of hypergastrinemia (eg, achlorhydria). When the diagnosis is in question, the secretin stimulation test, which stimulates gastrin secretion from gastrinomas but not from normal tissue, may be employed. Note: The availability of secretin for parenteral administration is now quite limited. Histamine receptor blockers and proton pump inhibitors reflexively increase serum gastrin levels and should be discontinued 30 hours and 7 days, respectively, prior to testing to exclude gastrinoma. Others have advocated measurements of multiple gastrointestinal hormones following a standardized mixed meal as the most efficient means of detecting the presence of neuroendocrine tumors in MEN1.
Because of their small size, gastrinomas can be difficult to localize in MEN1. Computed tomography (CT) and magnetic resonance imaging (MRI) may be useful in identifying larger lesions but are typically not helpful in identifying smaller ones. These imaging procedures are useful, however, in demonstrating hepatic metastases when present. The most promising localization techniques studied to date include endoscopic and intraoperative ultrasound, selective arterial secretin injection (followed by hepatic vein sampling for gastrin), and radiolabeled octreotide scanning. Each of these has been employed successfully to identify tumors in studies involving small groups of patients. It is important to recognize, however, that almost half of gastrinomas are not found with preoperative localization studies.
Insulinomas in MEN1 are detected using conventional biochemical testing (see Chapter 18). They can also be difficult to localize given their potential for multicentricity. Endoscopic ultrasound and selective arterial infusions of calcium with hepatic vein sampling (for insulin) have been used successfully to identify lesions in small groups of patients.
Glucagonomas, VIPomas, and somatostatinomas are diagnosed based on two fold or greater elevations in plasma glucagons, VIP, or somatostatin levels, respectively, in the presence of one or more pancreatic nodules. Nonfunctional tumors are typically diagnosed following imaging studies after exclusion of elevated levels of the plasma secretory products described above, or, more rarely, following measurement of pancreatic polypeptide levels.
Pituitary adenomas occur in approximately 25% of patients harboring the MEN-1 gene. The majority secrete prolactin, with or without secretion of excess growth hormone, followed by those secreting growth hormone alone, nonfunctional tumors, and those secreting excessive amounts of adrenocorticotropic hormone (ACTH) (Cushing disease). A prolactinoma variant of MEN1 has been described (Burin variant). This variant is characterized by an increased frequency of prolactinomas, carcinoids, and hyperparathyroidism and infrequent appearance of gastrinomas in affected kindreds. It does not appear to be associated with a specific mutation of the MEN1 gene. In one instance it has been linked to a germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1, but this has not been confirmed in other patients. Pituitary tumors in MEN1 are rarely malignant, but recent studies suggest that they may be larger and more aggressive than their sporadic counterparts. There is a high incidence of macroadenoma, they display a propensity for making more than a single pituitary hormone, and there is a suggestion that these tumors do not respond as well to medical therapy as do their sporadic counterparts. Diagnosis and management are similar to that of their sporadic counterparts (see Chapter 4).
Adrenal adenomas, including cortisol-producing adenomas, are seen in MEN1, theoretically making the differential diagnosis of Cushing syndrome in this setting complex (ie, adrenal adenoma vs basophilic adenoma of the pituitary gland vs ectopic ACTH secretion from a carcinoid tumor, which is also commonly associated with this syndrome). Empirically, most hypercortisolemia in this setting is due to pituitary disease. Adrenal adenomas are often found together with islet cell tumors in affected patients, and at least in some series they appear to lack the MEN1 genetic defect. This has led to the suggestion that they represent a secondary rather than a primary manifestation of the underlying genetic defect. This, however, remains controversial. Pheochromocytomas harboring an MEN1 rather than an MEN2 mutation have been described in seven patients. Thyroid disease has been said to be more common in MEN1; however, with the possible exception of thyroid adenomas, this link remains obscure. Subcutaneous lipomas, skin collagenomas, and multiple facial angiofibromas (particularly on the upper lip and vermillion border of the lip) are seen in 30% to 90% of family members in affected kindreds. Although clinically of little importance, when present, they may prove useful in identifying affected individuals within a kindred and lead to more effective screening (discussed later). Carcinoid tumors are seen with increased frequency in MEN1. They are almost exclusively foregut carcinoids and may be found in the thymus, in the lung (bronchial carcinoids), or in the gastric mucosa. It is likely that chronic hypergastrinemia (eg, that resulting from coexistent ZES and proton pump inhibitor therapy) contributes to the increased incidence of gastric carcinoids in MEN1. For unclear reasons, thymic carcinoids appear more commonly in males, bronchial carcinoids in females. They occasionally secrete hormonal products (eg, ectopic ACTH), are often malignant, and may behave aggressively. Leiomyomas have rarely been described in MEN1. Disease-specific mortality is due to pancreatic islet carcinoma and malignant thymic carcinoid.
MEN1 is inherited as an autosomal dominant trait. Traditional linkage studies localized the defective gene to the long arm of chromosome 11q13. Parallel analyses of DNA from endocrine tumors taken from MEN1 patients demonstrated allelic loss in this area, frequently resulting from large DNA deletions. This raised the possibility that the defective gene was a tumor suppressor gene involved in the control of cellular growth. In this paradigm (Figure 22–1), the inherited defective allele is silent in the presence of a normal, functioning allele on the second chromosome. A subsequent somatic mutation (often a deletion that removes the normal allele) results in a null genotype in which the suppressor gene locus is either absent or defective on both alleles. The high frequency with which such deletions occur is thought to account for the dominant nature of this particular genetic defect. Release of the tumor suppressor gene’s growth regulatory activity results in a hyperplastic growth response in cells harboring the somatic mutation. This promitogenic state probably provides the substrate for subsequent somatic mutations that result in acquisition of a more aggressive, and at times malignant, phenotype, as occurs in gastrinomas associated with ZES. Recent studies have succeeded in identifying the gene, termed the menin gene, which appears to be responsible for MEN1 (Figure 22–2). Mutations have been identified throughout the entire 610-amino-acid length of the menin coding sequence and include nonsense mutations, missense mutations, and deletions. Over 400 independent mutations have been described in MEN1 kindreds to date. Nevertheless, 5% to 10% of presumed MEN1 mutations are not detectable using available methodology. Menin is a ubiquitously expressed tumor suppressor gene product which has been shown to interact with several components of the trithorax family histone methyltransferase complex, including the mixed lineage leukemia (MLL) proteins. Menin, through this complex, controls expression of the cyclin-dependent kinase inhibitors p27Kip1 and p18Ink4c. This complex also methylates the lysine 4 residue of histone 3 (H3K4), a modification which has been linked to transcriptional activation. Loss of function of menin (eg, through those mutations associated with MEN1) results in downregulation of p27Kip1 and p18Ink4c and deregulated cell growth. Menin also suppresses expression of the developmentally programmed transcription factor, HLXB9, which may also lead to increased growth in menin-deficient cells. Mice heterozygous for deletion of the menin gene locus (Men1+/−) develop parathyroid dysplasia, adenomas, and carcinomas; pancreatic islet tumors, anterior pituitary tumors, and adrenal cortical tumors, all of which display loss of heterozygosity at the Men locus.
Figure 22–1
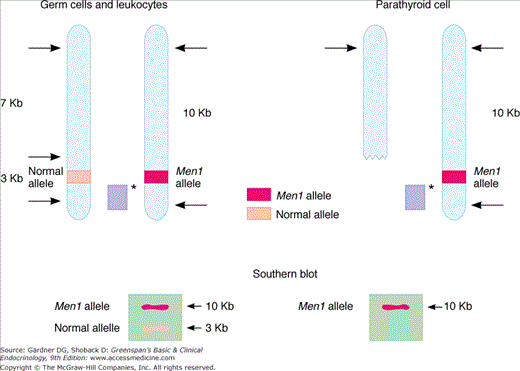
An example of allelic loss in Men1 involving large deletion of genetic material. Afflicted patient’s germ cells (shown on left) harbor both a normal and defective gene at the Men1 locus. Each of these is detected using selective restriction enzyme digestion and Southern blot analysis of the genomic DNA. Affected somatic cells (eg, parathyroid chief cells) undergo a second mutation, typically a deletion of the normal allele, resulting in detection of only the mutant allele by Southern analysis. Sporadic disease is thought to follow sequential mutation or deletion of each Men1 allele in the somatic cell. Solid bar (light purple) with asterisk represents radiolabeled probe used in Southern analysis; arrows indicate points of restriction enzyme digestion.
Figure 22–2
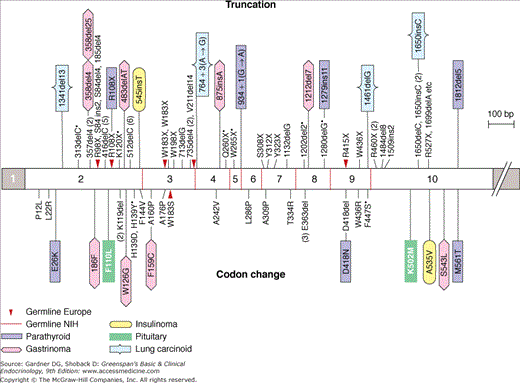
Some of the more common germline menin mutations from 56 MEN1 kindreds and somatic mutations from 24 endocrine tumors. Mutations above the bar cause protein truncations through stop codons or frameshifts leading to premature stop codons; two cause splice errors. Mutations below the bar cause missense or single-amino-acid codon changes.
(Reproduced, with permission, from Marx S, et al. Multiple endocrine neoplasia type 1: clinical and genetic topics. Ann Intern Med. 1998;129:484.)
The Men1 gene is also the gene most frequently mutated in a variety of sporadic endocrine tumors. Parathyroid adenomas (20%), gastrinomas (40%), insulinomas (15%), VIP-secreting tumors (60%), nonfunctioning pancreatic tumors (15%), glucagonoma (60%), adrenal cortical tumors (<5%), angiofibromas (10%), lipomas (30%), bronchial carcinoids (35%), and pituitary tumors (<5%) often harbor somatic mutations at the Men1 locus. It is important to note that screening for germline mutations at the Men1 locus fails to detect mutations 5% to 10% of the time despite evidence for loss of heterozygosity at 11q13. It is thought that the relevant mutations may be in regulatory regions surrounding the Men1 gene. Haplotype testing or genetic linkage analysis can be useful in these cases to identify relevant Men1 carriers. Unlike MEN2 (see later), there is no clear association between specific Men1 gene mutations and the nature or extent of endocrine gland involvement, although nonsense mutations at Tyr312 and Arg460 have been shown to be more common with the Burin variant.
Therapy of hyperparathyroidism in MEN1 is directed toward surgical extirpation of hyperplastic parathyroid tissue, typically with resection of three and one-half glands. This leaves one-half gland in situ in an attempt to preserve residual parathyroid function and avoid hypoparathyroidism. Alternatively, patients may be subjected to total parathyroidectomy with transplantation of the most normal appearing tissue to the nondominant forearm. There have been no head-to-head comparisons made between these approaches. Total parathyroidectomy (with transplant) is associated with a higher frequency of hypoparathyroidism, whereas subtotal parathyroidectomy has a higher rate of recurrence. Prophylactic near-total thymectomy is often performed at the time of neck exploration to remove potential intrathymic parathyroid glands as well as thymic carcinoid tumors.
Both persistence and recurrence of hyperparathyroidism occur more frequently in MEN1 than in sporadic disease. Persistence of hyperparathyroidism, defined as a failure to normalize serum calcium and parathyroid hormone (PTH) levels following the initial surgery, with removal of 3 to 3.5 parathyroid glands, occurs in 12% of cases of MEN1. Recurrent disease, defined as reappearance of hyperparathyroidism following at least 3 months of normocalcemia, is seen in as many as 44% of these cases 8 to 12 years following surgery. The high frequency of persistent hyperparathyroidism in MEN1 probably reflects the high frequency of supernumerary glands (prevalence as high as 30%) and ectopically located parathyroid tissue in patients carrying the Men1 gene. The increased frequency of recurrent disease is thought to result from the continued presence of the underlying mitogenic stimulus that drives parathyroid gland growth in this syndrome. Reoperation for recurrent disease in the neck is successful about 90% of the time, whereas autograft removal is successful in about 60% of patients.
Therapy of gastrinomas in MEN1 remains controversial. Suppression of gastric acid production with proton pump inhibitors (eg, omeprazole) remains a mainstay of therapy. Conservative medical management of these tumors has been predicated on their assumed low-grade malignant behavior, the dominance of complications related to gastric acid hypersecretion in contributing to morbidity and mortality, and the failure of most attempts at surgical resection to alter the natural course of the disease. More recently, recognition of the potential for more aggressive behavior in some of these tumors—in a recent study, 14% of these tumors demonstrated aggressive growth—and the fact that many of these tumors are ectopically located in duodenal submucosa and peripancreatic lymph nodes rather than the pancreatic bed has renewed interest in the possibility of surgical cure. A number of small studies have reported encouraging results when measures to both localize and remove gastrinoma tissue in the pancreas, duodenum, and regional lymph nodes have been used. Although the nature of the underlying genetic lesion and the multicentricity of these tumors may place limits on the prospects for cure of this disease in most patients, the slow growth characteristics of these tumors permit long periods of symptom-free survival following reduction of the tumor burden. For patients with liver or other metastatic disease, symptoms related to hypergastrinemia may be controlled with the proton pump antagonists, as described earlier. More conventional cancer therapy (eg, systemic chemotherapy, radiation therapy, or selective chemoembolization of hepatic metastases) is palliative and reserved for advanced stages of the disease. Islet tumors more than 3 cm carry an increased risk of malignancy and are usually resected regardless of functional status.
It is important to remember that calcium stimulates gastric acid secretion. This may occur through gastrin-dependent and gastrin-independent pathways. In MEN1 patients with both hyperparathyroidism and ZES, correction of the hyperparathyroidism and attendant hypercalcemia frequently results in a reduction in both basal and maximal acid output and a decline in serum gastrin levels. Secretin stimulation tests often normalize following parathyroidectomy. More importantly, there is a reduction in the dose of medication (eg, proton pump inhibitor) required to control symptoms of ZES following parathyroid surgery in approximately 60% of patients.
Unlike gastrinomas, insulinomas are rarely localized outside the pancreatic bed. Therefore, a more aggressive approach can be directed toward the pancreas in planning surgical resection. Enucleation of the identifiable lesions in the pancreatic head and blind resection of the pancreatic body and tail are often more successful in correcting hyperinsulinemia than in restoring normal gastrin levels in patients with ZES. Nonsurgical candidates (eg, those with serious coexisting disease or those in whom candidate tumors cannot be identified) can be managed with conventional medical therapy (eg, diazoxide or verapamil). (See Chapter 18.)
Nonfunctional tumors, the most common pancreatoduodenal neuroendocrine tumors in MEN1, are managed somewhat differently than the functional tumors. Surgical efforts to minimize spread of metastases are weighed against the need to avoid incurring surgical complications, including mortality, with little clinical benefit. In general, surgery is not recommended for tumors less than 2 cm in diameter. Life expectancy of MEN1 patients with nonfunctional tumors (∼43 years) is similar to that of those with gastrinomas and considerably shorter than those with other types of functional tumors (∼61 years).
MEN1 accounts for less than 1% of all pituitary tumors, 2% to 4% of cases of primary hyperparathyroidism, and about 25% of all gastrinomas. Thus, although routine screening for Men1 gene mutations is not indicated for sporadic cases of hyperparathyroidism or patients with pituitary tumors, screening of all cases of ZES is likely to be cost-effective in identifying carriers. The frequency of germline mutations in the Men1 gene in patients with tumors thought to be sporadic based on family analysis is about 5% for gastrinomas and 1% to 2% for other manifestations (eg, hyperparathyroidism, prolactinomas). Excluding those patients with ZES, screening for MEN1 outside affected MEN1 kindreds should be limited to those individuals for whom the index of suspicion for the syndrome is high (eg, familial history of endocrine tumors or hypersecretory states; history of multiple endocrine tumors or multigland involvement with hyperparathyroidism in the propositus). Identification of the carrier state should be performed with the intent of acquiring information that allows the clinician to focus screening on the relevant patient population (eg, members of an affected kindred who do not share the Men1 gene mutation need not be subjected to follow-up screening). Unlike the situation in MEN2 (see later), carrier analysis should not be used to support a major therapeutic intervention. Such interventions (eg, pancreatic exploration) may be associated with significant morbidity, and there is no evidence that they prolong patient survival. Patients with hyperparathyroidism should be screened for MEN1—even in the absence of a positive family history or any history of multiple endocrine tumors—if parathyroid hyperplasia is identified at the time of parathyroidectomy or if there is a history of recurrent hyperparathyroidism following parathyroidectomy. Patients with ZES should be screened for MEN1 given its high frequency (∼25%) in such individuals. Patients with isolated pituitary lesions have a low probability of having coexistent MEN1 and probably do not require screening unless there are other clinical features suggesting the syndrome. Genetic screening is available commercially at a number of locations (see www.genetests.org).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


